Day 1 :
Keynote Forum
Catherine GUILLOU
Unversité de Paris-Saclay, France
Keynote: New potent MKLP2 inhibitors in the paprotrain series with high anticancer activity in vivo : Design, Synthesis and Structure-Activity relationships
Time : 08:45- 09:15

Biography:
Research activities developed within Catherine Guillou’s group are centred on the development of new methodologies, their application to the total synthesis of biologically active natural products and the design of enzyme inhibitors involved in cancer and Alzheimer’s disease. She is co-authors of 58 publications and 8 patents.
Abstract:
Kinesins constitute a superfamily of microtubule-based motor proteins with important cellular functions ranging from intracellular transport to cell division. Some kinesin family members function during the mitotic phase of the eukaryotic cell cycle and are crucial for the successful progression of cell division. MKLP-2 (also known as Kif20A, RabK6, RB6K, Rab6KIFL, Rabkinesin6) a member of the kinesin-6 family plays an essential role during cytokinesis and is overexpressed in various cancers such as pancreatic cancer, bladder cancer, breast cancer, small-cell lung cancer, hepatocarcinogenesis, melanoma and gastric cancer. Furthermore, MKLP-2 is weakly detectable or absent in the normal spleen, lymph nodes, pancreas, lung, brain, liver, kidney and skeletal muscle. MKLP2 is involved in the relocation of chromosome passenger complex CPC (which consists of Aurora B, INCENP, surviving and borealin) to the spindle midzone. Down regulation of MKLP2 inhibits the growth of gastric and pancreatic cancer cells. We recently identified one compound (named paprotrain) as the first known inhibitor of MKLP-2. Paprotrain does not inhibit others members of the kinesin superfamily involved in mitosis. The synthesis, the structure-activity relationships and the biological activities will be discussed.
Keynote Forum
Mark McLaughlin
Merck Sharp & Dohme (MSD) Dept. of Process Research and Development, USA
Keynote: Mutagenic Impurities in Pharmaceuticals: ICH M7, Purge Factors and the Mirabilis Project
Time : 09:15 - 09:45

Biography:
Dr. Mark McLaughlin received his Ph.D from Strathclyde University under the joint supervision of Prof. Kerr and Prof. Pauson. He completed two post-doctoral appointments at the University of California, Berkeley with Prof. Rapoport and Prof. Heathcock. He received further education at the ETH, Switzerland (Prof. Diederich) and additional industrial experience at GSK (medicinal chemistry) and Astrazeneca (process chemistry). He joined MSD in 2003 as a Senior Research chemist and is currently a Principal Scientist. In addition to project leader responsibilities for the development of novel chemical processes supporting new drug applications, Mark has served as a “subject matter expert” within the Process Chemistry Dept. at MSD on the topic of mutagenic impurities. Contact: mark_mclaughlin@merck.com; Merck Research Labs PO Box 2000, RY800-C269 Rahway, NJ 07065.
Abstract:
Mutagenic impurities (MIs) are a special category of impurities that can be present in active pharmaceutical ingredients (APIs). The understanding, detection, and control of MIs have received increasing industry and regulatory attention over the past decade. Originated by Teasdale in 2010,1,2 the concept of “purge factor calculation” as a means to understand the fate of MIs in synthetic processes has been validated by various independent groups, including its application to a development project at MSD that was described in a recent publication.3 This approach can reduce the burden of analytical testing for MIs without compromising patient safety, provided a scientifically rigorous approach is taken, backed up by sufficient theoretical and/or analytical data. Moreover, specific reference to this method is provided in the accepted regulatory guidance – ICH M7 Option 4 - which was released in 2014. This presentation provides some background to the concept of purge factor calculation and introduces a consortium-led initiative, the Lhasa Mirabilis Project.4 The Mirabilis consortium seeks to develop an industry-standardised approach, providing an expert and scientifically robust software for the automated calculation of purge factors for potentially mutagenic impurities in a synthetic route.
Keynote Forum
Gervais Bérubé
Université du Québec à Trois-Rivières, Canada
Keynote: From para-aminobenzoic acid to small compounds with multiple biological properties
Time : 09:45 - 10:15

Biography:
Gervais Bérubé completed his Ph.D. in 1986 in the field of Organic Chemistry at the Université de Sherbrooke. After completing three post-doctoral stays initially in the field of Organic Chemistry, then in Oncology and lastly in Immunology, he became assistant professor of Medicinal Chemistry in the School of Pharmacy, Memorial University of Newfoundland. He is now professor of Organic Chemistry at the Université du Québec à Trois-Rivières. His main research interest is the design and development of new anticancer drugs. He has published more than 76 papers in reputed journals and presented more than 110 communications in diverse meetings.
Abstract:
The discovery of new molecules possessing multiple biological properties in a single entity is a subject of great interest to the scientific community. A simple compound previously used in our laboratory as a heterobifunctional linker to construct immunoconjugates was identified as a potential lead drug showing anti-inflammatory, anti-metastatic as well as anticancer activities. The lead compound is made in only three chemical steps from para-aminobenzoic acid with 43% overall yield. Its structure can be modified to give alternative analogs with similar activities. The prospect of lead optimization is significant. This communication will highlight the chemical and biological potential (in vitro and in vivo) of these type of molecules tested on bladder cancer. Amongst the results, the lead compound can reduce the size of a tumor in an animal model by 90% within 25 days without apparent side effects.
Keynote Forum
Afzal R Mohammed
Aston University, UK
Keynote: Dry Particle Coating for Pharmaceutical Applications: Opportunities, progress and current developments
Time : 9:25- 9:50

Biography:
Professor Afzal Mohammed holds a personal Chair in Pharmaceutics and has research expertise in paediatric medicines development which includes pharmaceutical excipient characterization, process and technology innovation to develop orally disintegrating tablets. He has over 15 years of experience in developing novel pharmaceutical processes and formulations and is the lead inventor for dry particle coater. He has over 100 publications including research articles, review papers, book chapters and patent applications.
Abstract:
Dry powder coating is a micro particle engineering process and involves the adsorption of “guest” particles onto the surface of “carrier” particles. This process requires conditions that enable frequent contact between guest and carrier particles, particle adsorption, and ultimately lowering of surface energy of the binary mixture. Although the theoretical paradigm for dry-coating is known, progress and pragmatic translation have been limited owing to the lack of processes and devices capable of producing composite particles while maintaining the innate characteristics of both components (guest and carrier). For instance, mechanofusion works on the principle of high centrifugal forces that generate heat thereby limiting its pharmaceutical application to heat labile materials. Similarly, processing materials using hybridiser can lead to particle attrition. One of the distinct advantages of this technology is the cross application of the fundamental principles to develop solutions for a wide range of different problems. For instance understanding the role of surface texture of carrier particles on the strength of interaction between the guest and carrier particles can provide vital information on its impact on flowability, guest stability (as a composite particle) as well as functionality of the resultant particles. Research at Aston University within our group has led to the development of a dry particle coater which can produce micro functionalised particles. We have characterised the resultant particles using range of techniques including AFM, surface interferometry, confocal microscopy, inverse gas chromatography and particle size analysis. The resultant particles were studied for flowability enhancement, content uniformity and micro particle based modified drug release.
Keynote Forum
Rongshi Li
University of Nebraska Medical Center, USA
Keynote: Fragment-based, Structure-guided and Natural Product-derived Small Molecules as Antibiotic and Anticancer Agents
Time : 9:50- 10:15

Biography:
Abstract:
- Computational Chemistry and Chemical Biology | Natural Products Chemistry | Pharmaceutical Analysis

Chair
Larisa Klapshina
Nizhny Novgorod State University, Russia

Co-Chair
Marco Marazzi
University of Lorraine-Nancy & CNRS, France
Session Introduction
Emmanuel Roulland
Université Paris Descartes, France
Title: Total Synthesis of Tiacumicin B Aglycone, a DFT-Guided Strategy

Biography:
Abstract:
Tiacumicin B is an antibiotic endowed with the remarkable ability to interact with a new biological target giving it an inestimable potential in the context of the ever-growing and worrisome apparition of resistances of bacteria and mycobacteria to antibiotics. The development of an efficient synthesis of this complex molecule will allow accessing valuable analogues. We have achieved the total synthesis of the tiacumicin B aglycone featuring the DFT-guided strategy concept. Macrolactone thus obtained is ready for subsequent glycosylation step. Starting from known alcohol (±)-3, this 16 steps synthesis was performed in a 3.6 % overall yield, only 4 steps dealing with the installation or the removal of protective groups. Relying on DFT predictions, we dared to use an unprecedented [2,3]-Wittig rearrangement of the propargyl ether of tertiary allylic alcohol to synthesis the most densely functionalized fragment of the target. We also accessed the tetrasubstituted C12-C15 diene stereoselectively using an innovative strategy based on the sequence allene-alkyne Pd/Cu-catalyzed cross-coupling / selective hydrosulfuration / Pd-catalyzed Kumada-Corriu cross-coupling of an alkenylsulfide function. To end this synthesis the E configuration of the C4=C5 bond was controlled thanks to a selective cross-metathesis of vinylborate, and a Suzuki cross-coupling was used to install the missing C1-C3 fragment. The final macrolactonization step was found to be ring-size-selective as again predicted by DFT.
Aramice Y. S. Malkhasian
King Abdulaziz University, Saudi Arabia
Title: Automated Drug Design of Kinase Inhibitors to treat Chronic Myeloid Leukemia

Biography:
Dr. Aramice Y. S. Malkhasian completed Ph.D. at Concordia University 1985and did Research scientist at McGill with M. A. Whitehead at 1995, at 2003 and 2004 work with Prof Michael Sevillaand Prof. Ferman Chaves at Oakland university. Currently professor at King Abdulaziz University
Abstract:
Medicinal chemistry has in the past been dominated by learned insights from experienced organic chemists. However, with the advent of computer based methods, computer aided drug design has become prominent. We have compared here the ability of expert chemists to purely automated methods and found that the automated method produces a better potential candidate drug than the expert input. The example chosen is based on inhibitors to Abl-kinase and the successful anti-leukaemic drug imatinib. The proposed molecule is a simple modification of nilotinib and has a docking energy of 4.2 kJ/mol better than the best intuitive molecule
Marco Marazzi
University of Lorraine−Nancy & CNRS, France
Title: DNA photosensitization: irreversible lesions caused by non-covalent binding with organic dyes

Biography:
After completing a PhD at the University of Alcalá, Madrid (Spain) in 2013, Marco Marazzi was a Humboldt Fellow at the Karlsruhe Institute of Technology (Germany). Starting from 2015, he is a researcher at the French National Center for Scientific Research (CNRS) and at the University of Lorraine in Nancy (France). Always interested in photoinduced processes of biologically relevant systems, he studied especially organic molecular switches applied to peptide conformational changes, channelrhodopsin as an optogenetic tool, and DNA photosensitization via Type I and II processes. His expertise covers a broad range of modeling techniques.
Abstract:
Although diverse DNA photostability mechanisms exist, organic dyes in the vicinity of DNA can induce damages through indirect light absorption. Hence, understanding the underlying mechanisms involved in photosensitized DNA damage is crucial to describe and possibly anticipate photobiological risks, as well as to design anticancer phototherapies. Here, we present the results concerning our latest studies on different reactivities induced by common organic dyes, through multiscale molecular modeling techniques coupled to spectroscopy experiments. Especially, the photochemistry of benzophenone – a paradigmatic DNA photosensitizer – and its implications in the competitive processes of hydrogen abstraction and energy transfer to DNA will be described. Electron transfer is also considered by the interaction of DNA with two fluorescent dyes widely used in cellular biology: nile blue and nile red. Finally, the potentialities of a very recently sinthesized novel carbazole in photosensitizing DNA through two-photon absorption will be reported. Especially, it will be shown how it can induce DNA strand break upon photoionization with the production of a solvated electron. The main advantage is the low-energy (infra-red) irradiation required also in the absence of molecuar oxygen, i.e. a prodrug of great interest for the potential treatment of solid tumors.
Alvaro Escribano
Universidad Carlos III de Madrid, Spain
Title: Patent propensity, R&D and market competition: Dynamic spillovers of innovation leaders and followers

Biography:
Abstract:
Sundaram Singh
Indian Institute of Technology (BHU), India
Title: An Efficient One Pot Multicomponent synthesis Of Some Novel Substituted Imidazoles using Nano Zirconia Catalyst Under solvent Free Conditions: A Greener “NOSE†approach

Biography:
Dr sundaram singh has completed her PhD at the age of 30 years from BHU, VARANASI . She is the Associate Professor of Chemistry Deptt, IIT(BHU),Varanasi. She has published more than 20 papers in reputed journals. Her research area is green synthesis, organic synthesis and evaluation of biological activity.
Abstract:
An increase in regulatory limitations on the use, manufacture and disposal of perilous organic solvents has focused attention on the development of non-hazardous alternatives such as solvent-less synthesis, multicomponent reactions and reusable heterogeneous catalysts for the sustainable development of chemical enterprise. These organic reactions possess many advantages over traditional reactions in organic solvents. For example, solvent-less, multicomponent reactions with reusable heterogeneous catalysts reduce the consumption of environmentally hazardous solvents and minimize the formation of other waste. The reactions occur under mild conditions and usually require easier workup procedures and simpler equipment.
Nano zirconia (ZrO2) have been widely investigated in the past decades due to their multiple potential applications. The crystal phase of ZrO2 (monoclinic and tetragonal) strongly influences the catalyst activities and selectivities.
A highly efficient method for the synthesis of substituted imidazoles from a multicomponent reaction of isatin derivatives with ammonium acetate and aromatic aldehydes under solvent free conditions has been established. The reaction is supposed to proceed via nano ZrO2-catalyzed C=O bond activation followed by the formation of diamine intermediate and its condensation with ZrO2 activated isatin derivatives. Because of the simple and readily available starting materials, easy operation and high bioactivity of imidazoles, this strategy can be broadly applied to medical chemistry. The recyclability of the nano ZrO2 catalyst is another emphasis of proposed methodology.
Scheme 1: Synthesis of imidazole derivatives 4a-r via multicomponent reaction of isatin derivatives 1a-f with ammonium acetate 2 and substituted aromatic aldehydes 3a-f.
Shaaban K. Mohamed
Manchester Metropolitan University, UK
Title: Towards improving in drug delivery using Silver nano-techniques

Biography:
Shaaban K. Mohamed has completed his PhD at the age of 32 years from Minia University and postdoctoral studies from Didsburg University School of Chemistry, Germany and Manchester Metropolitan University, UK.. He is member of RSC and received the Knowlege Exchange award 2013, MMU, UK . He has published more than 260 papers in reputed journals and has been serving as an editorial board member of repute journals such as International Journal of Chemistry and Pharmaceutical Research, Greener Journal of Pharmacy and Pharmacology, and Journal of Pharmaceutical and Applied Chemistry.
Abstract:
The emergence of multidrug resistant (MDR) bacteria has necessitated the development of novel groups of antibiotics that effectively block or subvert bacterial growth [1,2]. It has been reported that different efforts and diverse investments have been made to develop novel strategies for improving the concept of antibiotic delivery that could enhance the limited activity of those vital antibiotics against such types of bacteria [3,4]. In the present study, amoxicillin trihydrate and Neomycin sulphate were used for the first time as both reducing and capping agents in synthesis of silver nanoparticles (AgNPs). The synthesized AgNPs were evaluated for their antibacterial and synergistic activity with antibiotics against selected human pathogenic bacteria.
Sangeeta Jagtap
PDEA’s Baburaoji Gholap College, India
Title: Use of Curcumine, a Pharmaceutically Useful Compound in Organic Reactions

Biography:
Sangeeta Jagtap received her M.Sc.in Organic Chemistry and is a recipient of Gold Medal from Bombay University (1992), India. She completed her Ph.D. from National Chemical Laboratory, Pune, India, under the guidance of Dr. R. M. Deshpande. She worked as a visiting scholar at Stanford University, California, USA, under the guidance of Prof. Barry Trost. She has also completed a ‘Post Graduate Diploma in Industrial Program in Pharmaceutical chemistry and Production’ and has cleared many competitive examinations like NET, SET, GATE and the tests organized by BARC. Presently she is working as Associate Professor at Baburaoji Gholap College, Sangvi, Pune, India. Her research interests are methodology, organic synthesis and catalysis. She has publications in reputed journals like Org. Lett., Tet. Lett., Cat. Today etc. She has authored a book named ‘Pharmaceutical, Medicinal and Natural Product Chemistry’.
Abstract:
Curcumin, mainly isolated from turmeric, for long is known for its anti-inflammatory and antioxidant activity. It is also known to be effective against neoplasms, Alzheimer's disease, Hodgkin's disease. There are large numbers of references available in literature about its biological activity and mode of action etc. Contrary to the popular belief that turmeric to be used only in household or as a medicine, we found an entirely different dimension to the whole idea of its activity. Curcumin is found to be active as catalyst. When complexed with palladium, it acts as a catalyst for coupling reactions. It catalyzes efficiently the Heck and Suzuki reactions almost upto 90-95%. The respective products have been isolated in good yield. Thus Curcumin is found to act as an efficient ligand in catalysis study.
- Pharmaceutical Chemistry Novel Aspects | Prospectives of Medicinal Chemistry | Drug Designing Methodologies | Pharmaceutical Analysis

Chair
Francisco Alonso
Universidad de Alicante/ Instituto de Síntesis Orgánica, Spain

Co-Chair
Alexander V. Sirotkin
Constantine the Philosopher University and Research Institute of Animal Production, Slovakia
Session Introduction
Francisco Alonso
Universidad de Alicante, Spain
Title: Copper nanoparticles in organic synthesis

Biography:
Francisco Alonso received his B.Sc. (1986), M.Sc. (1988), and Ph.D. (1991) degrees in Chemistry from the University of Alicante (Spain). After a postdoctoral stay (1992–1994) at the University of Oxford (UK) with Prof. S. G. Davies, he moved back to the University of Alicante, where he is Full Professor of Organic Chemistry, Director of the Instituto de Síntesis Orgánica (ISO) and coordinator of the doctorate programme “Organic Synthesis”. He has authored 115 manuscripts, 3 patents and several book chapters, being co-founder of Medalchemy S. L. His research is focused on the development of new synthetic methodologies involving transition-metal nanoparticles and on the synthesis of biologically active natural or synthetic molecules. He is a member of the advisory board of Current Green Chemistry.
Abstract:
Some years ago, we developed a catalyst consisting of copper nanoparticles on activated carbon (CuNPs/C) which was shown to be very versatile in the synthesis of 1,2,3-triazoles through click chemistry.1 More recently, we have effectively accomplished the multicomponent synthesis of indolizines using 0.5 mol% CuNPs/C as catalyst in dichloromethane.2 Interestingly, the same procedure, when applied in the absence of solvent using piperidine as the secondary amine, has led to heterocyclic chalcones with exclusive Z stereochemistry. The aforementioned copper-catalyzed three-component synthesis of indolizines, when followed by heterogeneous catalytic hydrogenation, allowed the straightforward preparation 1-dialkylamino-3-substituted indolizidines with high chemo- and diastereoselectivity, though an overall atom-economy protocol.3 Copper nanoparticles on zeolite Y has been found to be an effective catalyst for the cross-dehydrogenative coupling of tertiary amines and terminal alkynes (producing propargylamines) in the presence of tert-butyl hydroperoxide as the oxidant, without the need of an inert atmosphere and in the absence of solvent, using 1.5 mol% catalyst.4 The catalyst is reusable and more efficient than an array of commercial catalysts. All types of compounds presented, 1,2,3-triazoles, indolizines, chalcones, indolizidines and propargylamines, are of pharmacological interest, some of which have shown in-vitro prominent activity.
Mauro Safir Filho
Universite Côte d'Azur, France
Title: Rational Design and Synthesis of Innovative RNA Ligands to Target HCV Internal Ribosome Entry Site

Biography:
Mauro Safir Filho has a degree in Industrial Chemistry and a master’s degree in Chemistry from the Federal University of Rio Grande do Sul (UFRGS), Brazil. Now he is PhD student at University Côte d'Azur, in Nice, France. He has experience in organic synthesis and photochemistry and now has been working on methodologies of post-synthetic modifications of oligonucleotides.
Abstract:
Targeting RNA by using small molecules is one of the most intriguing challenges of current medicinal chemistry because, even if a large number of RNA-binding agents have already been identified, the rational design of synthetic molecules that would be specific for a particular RNA structure remains extremely difficult. Recently, this research field raises to an even greater interest since the discovery of new roles of non-coding RNA molecules, including the regulation of a wide number of biological processes as gene expression, tumorigenese and viral translation in chronic diseases, making them potential and important druggable targets. In this context, our research group devoted a lot of effort to develop small-sized organic molecule targeting RNAs. Our ligand design consisted in the combination of molecular recognition elements to enhance site specificity with electrostatic interactions to strengthen the complex stability. The preparation of these multimodal ligands was accomplished by assembling artificial nucleobases, able to form triplets through Hoogsteen interactions with A:U and G:C base pairs, with basic amino acid residues. The affinity and specificity of our ligands were evaluated towards the IIId loop of HCV Internal Ribosome Entry Site (IRES) as RNA model. Low micromolar dissociation constants could be obtained for our best ligand with two-fold higher affinity compared with the non-specific RNA binder neomycin used as positive control. Furthermore, high site specificity to target the single U:A base pair besides the bulge was also observed. Moreover, great selectivity to target the HCV IRES IIId loop instead natural tRNA was achieved.
This work is supported by CAPES (fellowship to MSF, process number 99999.0011495/2015-01)
Hamid Irannejad
Mazandaran University of Medical Sciences, Iran
Title: Synthesis of 5,6-diaryl-1,2,4-triazine derivatives with ethyl acetate moiety as novel neuroprotective agents against H2O2 and Aβ-induced neurotoxicity

Biography:
Hamid Irannejad has completed his Doctor of Pharmacy at Kerman University of Medical Sciences and PhD at Tehran University of Medical Sciences, Iran. His Postdoctoral studies were accomplished at University of Siena, Italy, under the supervision of Prof. Maurizio Botta. Currently, he is serving as an Assistant Professor at Mazandaran University of Medical Sciences. He has published nearly 20 papers in reputed journals in the field of Medicinal Chemistry.
Abstract:
Alzheimer’s disease is a neuropathologic disorder characterized by intracellular neurofibrillary tangles and amyloid aggregates in the CNS. In recent years numerous approaches have been used to combat AD like small molecule inhibitors of Aβ aggregation, anti-inflammatory agents, cholinesterase, β- and γ-secretase. Herein, we report synthesis of some 5,6-diaryl-1,2,4-triazines 3a-f and 8a-e as potential agents for treatment of AD. We evaluated them against both H2O2 and β-amyloid induced toxicity in PC-12 and SH-SY5Y cells and the extent of cell viability and apoptosis were assessed. The synthesis of compounds (3a-f) was started by 1,2-diketones, in which triazine ring closure was performed by thiosemicarbazide and alkylation by ethyl chloroacetate to afford compounds 3a-f. Synthetic route for compounds 8a-e was started by an acylation reaction of anisole with phenylacetic acid derivatives. The oximation in the alpha position of carbonyl group was performed by use of sodium methoxide and butylnitrite. The next two steps, were performed similarly to afford final compounds 8a-e. All compounds showed significant neuroprotective activity with EC50 values ranging from 14-30 µM. Most compounds could increase cell viability compared to amyloid treated group. Surprisingly, 3-thioxo-1,2,4-triazin-2(3H)-yl) acetate derivative 8e was the most potent compound in both tests with EC50 of 14 µM and could increase 40% of cell viability revealed by cytometric analysis with Annexin V/PI staining. It was also shown that 8e has more neuroprotective activity than Quercetin. Morphologic evaluation of cells by DAPI staining and TUNEL assay showed the effectiveness of this compound to improve neurite outgrowth in neuronal cells.
Victor Guallar
Barcelona Supercomputing Center, Spain
Title: PELE Studio: the next generation drug design software

Biography:
Guallar completed his PhD in collaboration between UAB (Spain) and UCBerkeley (USA) and postdoctoral studies from Columbia University. After an assistant professor position at Washington University School of Medicine (USA), he was awarded an ICREA professor position at the Barcelona Supercomputing Center. He is also founder of Nostrum Biodiscovery. He has published more than 100 papers in reputed journals and has been the recipient of presitioug grants like an Advanced ERC from the European Union.
Abstract:
We are clearly assisting a rise of computational predictions in drug design; top pharmaceutical companies are signing large contracts with modeling software companies. This, has been made possible by the improvement in the techniques (algorithms) but also by the rise in computational power, such as the use of graphic processing units (GPUs) in molecular dynamics of docking program. Next generation drug design software, however, will embrace additional (current) technological developments. In this line we are combining PELE, our Monte Carlo sampling technique highlighted as an outstunding achievement in the latest CSAR blind test, with machine learning algorithms, high performance computing and improved 2D/3D visualization techniques. Our aim is to provide a drug design software capable of: i) INTERACTIVE, instantaneous answers; ii) ACCURATE, quantitative answers; iii) SMART: self-learining capabilities; iv) CONNECTING: providing a virtual working space. Our efforts and initial results in this line will be provided in this talk.
Omer Abdalla Ahmed Hamdi
Alneelain University, Sudan
Title: Chemical constituents from the rhizomes of Curcuma zedoaria and assessment of their biological activities

Biography:
Omer Abdalla Ahmed Hamdi has completed his PhD from University Malaya, Malaysia. He is the director of Center of Natural Product Research and Drug Discovery,. He has published more than 12 papers in reputed journals and has been serving as supervisor for more than ten students for master and Ph.D program.
Abstract:
Phytochemical investigation of C. zedoaria resulted in the isolation of 21 compounds. Isolated compounds includes eighteen sesquiterpenes and three labdane diterpenes. Various chromatographic techniques were used for the detection and isolation of the compounds. Extensive spectroscopic methods including NMR, IR, UV, GC-MS, LC-MS were used for the identification of the isolated compounds. Isolated compounds were subjected to cytotoxicity, anti-oxidant and neuroprotective assays. Curcumenol and dehydrocurdione showed the highest protection (100%) against hydrogen peroxide induced oxidative stress in NG108-15 cells at the concentrations of 4 and 8 µM, respectively. In the oxygen radical antioxidant capacity assay, zerumbone epoxide showed the highest antioxidant activity with a Trolox equivalent (TE) of 35.41 µM per 100 µg of sample. In the MTT based cytotoxicity assay against four cancer cell lines (Ca 41 Ski, MCF-7, PC-3 and HT-29), curcumenone and curcumenol displayed strong antiproliferative activity (IC50 8.3 and 9.3µg/ml, respectively). A quantum chemical study was performed to investigate their relationship with cytotoxic activity and revealed that the dipole moment (µ), molecular volume (V), molecular area (A), polarizability (α) and hydrophobicity (log P) are the most important descriptors that influence the cytotoxic activity of the compounds under investigation. The two most active compounds; curcumenol and curcumenone were investigated for their binding to human serum albumin (HSA). The spectroflurometric analysis, in conjunction with molecular docking study suggested that both curcumenol and curcumenone could bind to binding sites I and II of HSA with intermediate affinity while site I was the preferred binding site for both molecules.
Sartaj Tabassum
King Saud University, Saudi Arabia
Title: Inorganic Pharmaceuticals : DNA-binding and anticancer activity of new compound

Biography:
Prof. Sartaj Tabassum is working as Professor in the Department of Chemistry, Aligarh Muslim University, Aligarh and Presently in King Saud University ,Riyadh Saudi Arabia He has published 105 papers in the journals of international repute. He is a life member of ICC, CRSI, ISCB, DNA Society of India and American Nano Society. He has successfully guided 16 Ph.D. He has successfully completed many research schemes granted by TWAS, Italy, CSIR, New Delhi, DBT, Govt. of India. As a distinguished Scientist, Prof. Tabassum was awarded Overseas Associateship award in 2005 by DBT, Govt of India. He has signed several MoU and joint research collaboration with University of Camerino UNICAM, Italy, USM Malaysia and USTC Hefei, China. He has visited many countries for academic pursuit particularly, China, USA, Italy,Saudi Arabia as fellow, visiting Professor and for the international conferences.
Abstract:
Medicinal inorganic pharmaceutical chemistry is an interdisciplinary thrust area of chemical biology research; is currently much more known for its many applications in enzyme mimic catalysis and also has enormous potential to act as therapeutic and diagnostic agents Development of new drug design and therapeutic strategies that could target cancer cells leaving normal cells unaffected still continues to be a challenge. Series of New pharmacophore of metallic compounds were designed, synthesized and characterized by various spectroscopic methods (IR, ESI–MS, 1H, 13C and Sn119 NMR) and further confirmed by X–ray crystallography. In vitro DNA binding studies of the compounds investigated by absorption and emission titration methods which revealed that 1 recognizes the minor groove of DNA in accordance with molecular docking studies with the DNA duplex. Gel electrophoretic assay demonstrates the ability of 1 to cleave pBR322 DNA through hydrolytic/oxidative process which were further validated by T4 religation assay. To understand the drug–protein interaction of which ultimate molecular target was DNA, the affinity of compounds towards HSA was also investigated by the spectroscopic and molecular modeling techniques which showed hydrophobic interaction in the subdomain IIA of HSA. The SOD-like activity of the compounds was evaluated using a xanthine/xanthine oxidase assay, which showed SOD activity in the micro molar range for both the heterobimetallic complexes viz., (IC50) 0.082 μM. Furthermore, complexes showed high inhibitory activity against Topo-Iα at a concentration of 20 μM as IC50, suggesting that complex is an efficient DNA cleaving agent. In vitro studies on the anticancer activity against the HepG2 hepatocellular carcinoma cell line revealed that complexes have the capability to kill the chosen cancer cell, but the efficiency of few complexes are higher than the reported earlier . The mode of cell death induced by complex is primarily apoptosis as revealed by AO/EB staining, Hoechst 33258 staining, and assessment of the mitochondrial trans-membrane potentia
Mandana Behbahani
University of Isfahan, Iran
Title: Inhibition of immunodeficiency type-1 virus (HIV-1) life cycle by magnetic water

Biography:
Abstract:
In the present study, the anti-HIV-1 activity of magnetic water at different intensities of 250, 750, 1000, 2000 gauge has been determined. A time of drug addition assay was done to identify the target of anti-HIV-1 agents. MW at different intensities showed potent anti-HIV-1 activity. EC90 of MW was achieved at intensity of 2000 gauge. The time of drug addition study demonstrated that the inhibitory effect of MW is before HIV-1 infection. The frequency and intensity of CD4, C-C chemokine receptor type 5 (CCR5) and chemokine receptor type 4 (CXCR4) on CD4+ T cells were not changed in cells treated with MW at different intensities. The results demonstrated that MW might be a suitable candidate for in vivo testing of anti-HIV infection.
Fatemeh Majidi Arlan
University of Urmia, Iran
Title: Synthesis of new series of pyrazolo[3,4-b][1,6]naphthyridine in presence of nano-Al2O3

Biography:
Fatemeh Majidi Arlan was born in Urmia (Iran) in 1984. She received his degree in Pure Chemistry from the Urmia University. She will receive his Ph.D degree in Organic Chemistry from the Urmia University (Iran) in 2017 after completing his research in the Study of Synthesis of chromene and pyridine heterocyclic derivatives by one-pot, multicomponent reaction of aryglyoxals, methylene active compounds and enamines in the presence of nanocatalysts and study of their interactions with heavy metals using analytical techniques under the guidance of Professor Jabbar Khalafy. Her current research interest focus on the Synthesis of new series of pyrazolo[3,4-b][1,6]naphthyridine in presence of nano-Al2O3
Abstract:
Pyrazolonaphthyridine is a fused polycyclic heterocycles with four nitrogen-atoms, the pyrazolonaphthyridine derivatives have received much attention in recent years due to their wide biological and pharmacological activities such as potent phosphodiesterase 10A inhibitors [1], selective histamine 4 receptor antagonists [2] bombesin receptor subtype-3 agonists [3] and protein kinase inhibitors [4]. Herein, we report the one-pot threecomponent synthesis of new series of pyrazolo[3,4-b][1,6]naphthyridine in the presence of different catalysts such as L-alanin, p-TSA and nano Al2O3. Ease of purification of products, isolation of catalysts, using water/ ethanol as a green solvent, high yields and shorter reaction times in presence of nanocatalyst in comparison with other catalysts are the advantages of this procedure.
Arezou Ghahghaei
University of Sistan and Baluchestan, Iran
Title: Protective effect of green synthesis gold nanoparticles (AuNPs) from Pulicaria undulata on the amyloid formation in α-lactalbumin

Biography:
Arezou Ghahghaei completed her PhD from University of Australia (Wollongong). She is Biochemist, Associated Professor and Head of Department of Biology. Her research focuses on pharmaceutics effect on protein aggregation. She has published several papers in reputed journals.
Abstract:
The formation and deposition of protein fibrillar aggregates in the tissues is associated with several neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease. Nanoparticles possess an enormous surface area and are found to inhibit protein amyloid formation. Recently plant-mediated nanoparticles synthesis has drawn a great deal of attention because it is rapid, environmentally friendly, cost effective and it provides a single step technique for the biosynthetic processes and is safe for human therapeutic use. The aim of this study was to assess the effect of green synthesis AgNPs from Pulicaria undulata L. on the reduction of protein aggregation in reduced α-lactalbumin. The results showed that green synthesis AuNPs have the ability to prevent the aggregation of α-lactalbumin in a concentration-dependent manner. This inhibitory effect of AuNPs probably caused by decreasing the rate of fibrillation through surface absorbing of free monomeric peptides and prevent amyloid fibril formation. In fact, by increasing the concentration of AuNPs within a specified range, the adsorption and interaction between AuNPs and protein have increased and protein conformational changes and self-association decreased, thus amyloid aggregation is prevented. In the main, results of this study show that green AuNPs mediated by Pulicaria undulata L. has the capability in inhibiting amyloid fibril formation and can be used as a therapeutic approach in the treatment of amyloid disease such as Alzheimer disease.
Yuegang Zuo
University of Massachusetts Dartmouth, USA
Title: Determination of Glimepiride in Pharmaceutical Formulations Using HILIC

Biography:
Yuegang Zuo is a Full Professor and Director of Graduate Programs at Department of Chemistry and Biochemistry, University of Massachusetts Dartmouth. He received his B.S. degree in chemistry from Wuhan University in 1982, M.S. degree in environmental chemistry from the Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, in 1984, and PhD in environmental science from Swiss Federal Institute of Technology Zurich in 1992. Most of his recent research has focused on separation, identification and quantification of PPCPs and phenolic antioxidants in plants, pharmaceuticals, foods and the environment and examine their occurrence, sources, bioeffects and fate in the biochemsphere.
Abstract:
Glimepiride is one of the most widely prescribed antidiabetic drugs and contains both hydrophobic and hydrophilic functional groups in its molecules, and thus could be analyzed by either reversed-phase high performance liquid chromatography (HPLC) or hydrophilic interaction liquid chromatography (HILIC) [Zuo Y (2014) (Eds.), High-Performance Liquid Chromatography (HPLC): Principles, Procedures and Practices. Nova Science Publishers, Inc., New York, USA; Zuo et al., Saudi Pharmaceutical J., 2017]. In the literature, however, only reversed-phase HPLC has been reported. In this study, a simple, rapid and accurate hydrophilic interaction liquid chromatographic method was developed for the determination of glimepiride in pharmaceutical formulations. The analytical method comprised a fast ultrasound-assisted extraction with acetonitrile as a solvent followed by HILIC separation and quantification. The effects of various HILIC parameters on the separation and determination will be discussed in details at the presentation. The developed method has been successfully applied to determine the glimepiride contents in pharmaceutical formulations and human fluids.
Fabio Luiz Paranhos Costa
Federal University of Goias, Brazil
Title: Very fast and surprisingly accurate GIAO-mPW1PW91/3-21G//PM7 scaling factor for 13C NMR chemical shifts calculation

Biography:
Fabio Luiz Paranhos Costa has completed his PhD from University Federal of Rio de Janeiro and Postdoctoral studies from Univesity Federal Fluminense. He has published more than 20 papers in reputed journals.
Abstract:
In this work, we present a new GIAO-HDFT universal scaling factor (mPW1PW91/3-21G//PM7(I)) and a comparative study in which is investigated its ability to predict NMR 13C chemical shifts (δ) with high cost-effectiveness ratio. A set of 22 small molecules providing 27 different 13C δ determined in the gas phase was used for all scaling factors protocols: B3PW91/ccpVDZ//B3PW91/cc-pVDZ (II), B3PW91/cc-pVTZ//B3PW91/cc-pVDZ (III), B3LYP/6-311+(2d,p)//B3LYP/6-31G(d) (IV), mPW1PW91/6-31G(d)//PM7(V) , mPW1PW91/6-31G(d)//mPW1PW91/6-31G(d) (VI). Despite the calculation approximations the δ calculated at the GIAO-mPW1PW91/3-21G//PM7 using a simple relationship (δscal = 1.14. δcalc – 4.7, where δcalc and δscal are the calculated and the linearly scaled values of the 13C δ, respectively) were able to yield MAD and RMS errors as small as those obtained with other GIAO-HDFT with bigger basis sets (protocols (II) to (VI)). The robustness of the new protocol and its applicability to practical problems was evaluated by the calculation of the δ for two natural compounds with synthesis, biological and therapeutic interest: tryptanthrin (indolo[2,1-b]quinazoline-6,12-dione) and (-)- loliolide (7aR)-6-hydroxy-4,4,7a-trimethyl-6,7-dihydro-5H-1-benzofuran-2-one. For both compound, the 6 protocols presented good agreement with experimental data. Moreover, for the second compound, the new protocol performs even better than the 5 others. In conclusion, GIAOmPW1PW91/3-21G//PM7 linear regression obtained by using the experimental and the calculated data, is a very attractive tool as an alternative to more computationally demanding approaches, which are usually applied in order to achieve 13C NMR δ calculations.
- Computational Chemistry and Chemical Biology | Heterocyclic Chemistry | Pharmaceutical Analysis

Chair
Vladislav Yu Korotaev
Ural Federal University, Russia

Co-Chair
Daisuke Yamamoto
Kitasato University, Japan
Session Introduction
Daisuke Yamamoto
Kitasato University, Japan
Title: Manganese-catalysed hydroperoxidation of carbon–carbon double bonds using molecular oxygen present in air and hydroxylamine under ambient conditions

Biography:
Daisuke Yamamoto has completed his PhD in 2006 at Kitasato University (Prof. Toshiaki Sunazuka). Subsequently, he became a researcher at the Kitasato Institute (Prof. Satoshi ÅŒmura) and joined the group of Prof. Barry M. Trost at Stanford University, as a postdoctoral fellow. In 2008, he started his academic career as an Assistant Professor at Kitasato University. His research focuses on the development of redox reactions with attendant applications in biologically active molecules.
Abstract:
Developing a new methodology for transition metal-catalysed oxidation reactions has been extensively studied in the recent decade, and molecular oxygen is essentially recognised as an ideal oxidant. Despite developing several elegant oxidation processes involving molecular oxygen as a sole oxidant, methodologies for directly incorporating molecular oxygen into organic substrates remains a major challenge in synthetic chemistry. In continuation of our studies on manganese-catalysed oxidative reactions, we have found that manganese(III) acetylacetonate is a highly efficient catalyst for hydroperoxidation of carbon-carbon double bonds of enynes as well as styrene derivatices using N-hydroxyphthalimide, N-hydroxybenzotriazole or N-hydroxysuccinimide under mild reaction conditions. This reaction proceeded at room temperature through the direct incorporation of molecular oxygen present in air. The required catalytic loading of manganese(III) acetylacetonate is extremely low (generally 0.02–0.5 mol%, and a minimum of 0.001 mol%). On the basis of this knowledge, we recently reported a manganese-promoted oxidative cyclisation of unsaturated oximes to provide 4,5-dihydroisoxazoline alcohols. In addition, we applied our method to the synthesis of hydroxamic acid, which is a promising antitrypanosomal agent for the management of Chagas disease. In the presentation, we will also discuss the further studies.

Biography:
El-Sawy has completed her PhD at the age of 28 years from Al-Azher University and postdoctoral studies from National Research Centre, Cairo Egypt. She has published more than 34 papers in reputed journals. Current position: Professor Doctor of Organic Chemistry at Chemistry of Natural Compounds Department, National Research Centre (NRC).
Abstract:
Resistance to conventional chemotherapy, leads to the need for development of novel safe and effective cancer therapies with new mechanism of action. Anti-angiogenic drugs are major example of such newly developed targeted therapeutics .In cancer drug development arena, coumarin-type compounds have been reported to bosses marked cytotoxic activities, in addition act as novel angiogenesis inhibitors. In this respect,a new series of coumarin derivatives was synthesized starting from 2-oxo-2H-coumarin-6-sulfonyl chloride (1), 6-nitro-2-oxo-2H-coumarin-3-sulfonyl chloride (10) and 6-amino coumarin-2-one (19). Reaction of 1 or 10 with 2-cyanoacetohydrazide, 2`-acetyl-2-cyanoacetohydrazide or 3-amino-5-pyrazolone afforded pyrazoline derivatives. While reaction of1 or 10 with malononitrile followed by reaction with hyadrazine hydrate, urea, thiourea or guanidine led to the formation of pyrazole and pyrimidine derivatives. On the other hand, compound 19 on reaction with VilsmeierHaack reagent yielded the corresponding aldehyde20. Compound 20 under reaction with chlorosulfonylisocyanate afforded N-chlorosulfonyl amid which cyclized to give pyranobenzothiazine derivative 25. The tested compounds 4, 5, 8, 12, 13 and 14 were non-cytotoxic against hepatocellular carcinoma cells (HepG2) using MMT. These non-cytotoxic compounds were evaluated as anti-angiogenicagent. Collectively, our results indicate that, coumarin molecules 4, 5, 8, 13 and 14 can be utilized as lead compounds to develop potential non-toxic angiogenesis inhibitors and small molecular ligands to target (HepG2), which was in concomitant with molecular docking results. 1-Acetyl-5-amino-4-(2-oxo-2H-chromene-6-sulfonyl)-1,2-dihydro-pyrazol-3-one (4) considered a promising anti-angiogenic agent, where it exhibited MMP-dependent anti-migratory activity and down regulated CD105.

Biography:
Korotaev V. Yu. graduated from the Ural State University in 1993 and has completed his PhD in 1998. Currently he works at the Department of Organic Chemistry of the Ural Federal University as a senior researcher. He has published more than 65 papers in peer-reviewed journals. His area of scientific interests includes unsaturated nitro compounds, heterocyclic compounds and fluoroorganic chemistry.
Abstract:
The [3+2] cycloaddition of sodium azide and stabilized azomethine ylides to 3-nitro-2-(trihalomethyl)-2H-chromenes 1 and 2 was studied and cycloadducts 3‒8 were synthesized. The cytotoxic activity of D3-annulated chromane derivatives is discussed.

Biography:
Tayebeh Hosseinnejad received his BSc, MSc and PhD degrees from University of Tehran in 2001, 2003 and 2007 respectively. She completed his Doctoral thesis under supervision of Prof. Hassan Behnejad. She joined as an Assistant Professor in Alzahra University, Iran. Her research interests focuses on computational organic and organometallic chemistry and computational thermodynamics.
Abstract:
Halloysite nanotubes (HNTs), with general formula of (Al2(OH)4Si2O5.2H2O) possess high surface area, tubular morphology and high mechanical strength. In HNTs a monolayer of water separates the unite layers in HNTs. Moreover, the exterior and interior surfaces of HNTs are chemically different. Tetrahedral SiO4 groups are located in the outer surface while the octahedral gibbsite Al(OH)3 sheet forms the inner surface. Recently, functionalization of the surface of HNTs and immobilization of metal nano particles are considered as a potent method for modification of the features of HNTs and expanding their applications specially as an effective catalyst in the selective synthesis of an specific isomer of pharmacetual compounds. In continuation of our attempt to introduce computational modeling of structural, electronic and thermochemical properties of heterogeneous nanocatalysts to design the regioselective synthesis of 1,2,3-triazoles as pharmacetual compounds, herein, we present a novel heterogeneous catalyst based on functionalization of HNTs with (3-chloropropyl) trimethoxysilan, thiosemicarbazide and furfural and incorporation of copper NPs. Considering the importance of understanding the surface chemical and physicochemical properties of functionalized HNTs, we investigated the computational modeling of regioselective synthesis of disubstituted 1,2,3-triazoles. Strictly speaking, we concentrated on the quantitative description of structural and electronic features of interactions between copper NPs and thiosemicarbazide functionalized HNTs modified with furfural (denoted as HNTs-T-F) via density functional theory (DFT) and quantum theory of atoms in molecules (QTAIM) approaches. Then, we applied our computational modeling in the design of reaction path so that it can be led to the synthesis of an specific isomer of disubstituted 1,2,3-triazoles as pharmacetual compounds.
Gisela Brändén,
University of Gothenburg, Sweden
Title: Structural studies of the novel antibacterial target MraY and its interaction with the natural inhibitor compound tunicamycin

Biography:
Abstract:
The rapid increase of antibiotic resistance has created an urgent need to develop novel antibacterial drugs. I will describe the crystal structure of the promising bacterial target phospho-N-acetylmuramoyl–pentapeptide translocase (MraY) in complex with the nucleoside antibiotic tunicamycin. The structure reveals the mode of action of several related natural-product antibiotics and also gives an indication on the binding mode of the MraY UDP–MurNAc–pentapeptide and undecaprenyl-phosphate substrates.
(Hakulinen et al. Nature Chemical Biology, 13:265-267, 2017)
Wajaht Amin Shah
University of Kashmir, India
Title: Synthesis and screening of ursolic acid-benylidine derivatives as potential anti-cancer agents

Biography:
Dr. Wajaht Amin Shah has completed his Ph.D. in natural product chemistry. He has published papers in various international journals on natural product chemistry and synthetic chemistry. He has produced various doctoral and pre doctoral students under his supervision. He is presently working as Associate Professor in the department of chemistry university of Kashmir and is actively involved in guiding PG students and doctoral students.
Abstract:
Ursolic acid present abundantly in plant kingdom is a well-known compound with various promising biological activities including, anti-cancer, anti-inflammatory, hepatoprotective, antiallergic and anti-HIV properties. Herein, a library of ursolic acid-benzylidine derivatives have been designed and synthesized using Claisen Schmidt condensation of ursolic acid with various aromatic aldehydes in an attempt to develop potent antitumor agents. The compounds were evaluated against a panel of four human carcinoma cell lines including, A-549 (lung), MCF-7 (breast), HCT-116 (colon), THP-1 (leukemia) and a normal human epithelial cell line (FR-2). The results from MTT assay revealed that all the compounds displayed high level of antitumor activities compared with the triazole analogs (previously reported) and the parent ursolic acid. However, compound 3b, the most active derivative was subjected to mechanistic studies to understand the underlying mechanism. The results revealed that compound 3b induced apoptosis in HCT-116 cell lines, arrest cell cycle in the G1 phase, caused accumulation of cytochrome c in the cytosol and increased the expression levels of caspase-9 and caspase-3 proteins. Therefore, compound 3b induces apoptosis in HCT-116 cells through mitochondrial pathway.
- Fianl Rejected
Chair
Fianal
Session Introduction
Mark McLaughlin
Merck Sharp & Dohme (MSD) Dept. of Process Research and Development, USA
Title: Mutagenic Impurities in Pharmaceuticals: ICH M7, Purge Factors and the Mirabilis Project

Biography:
Dr. Mark McLaughlin received his Ph.D from Strathclyde University under the joint supervision of Prof. Kerr and Prof. Pauson. He completed two post-doctoral appointments at the University of California, Berkeley with Prof. Rapoport and Prof. Heathcock. He received further education at the ETH, Switzerland (Prof. Diederich) and additional industrial experience at GSK (medicinal chemistry) and Astrazeneca (process chemistry). He joined MSD in 2003 as a Senior Research chemist and is currently a Principal Scientist. In addition to project leader responsibilities for the development of novel chemical processes supporting new drug applications, Mark has served as a “subject matter expert” within the Process Chemistry Dept. at MSD on the topic of mutagenic impurities. Contact: mark_mclaughlin@merck.com; Merck Research Labs PO Box 2000, RY800-C269 Rahway, NJ 07065.
Abstract:
Mutagenic impurities (MIs) are a special category of impurities that can be present in active pharmaceutical ingredients (APIs). The understanding, detection, and control of MIs have received increasing industry and regulatory attention over the past decade. Originated by Teasdale in 2010,1,2 the concept of “purge factor calculation” as a means to understand the fate of MIs in synthetic processes has been validated by various independent groups, including its application to a development project at MSD that was described in a recent publication.3 This approach can reduce the burden of analytical testing for MIs without compromising patient safety, provided a scientifically rigorous approach is taken, backed up by sufficient theoretical and/or analytical data. Moreover, specific reference to this method is provided in the accepted regulatory guidance – ICH M7 Option 4 - which was released in 2014. This presentation provides some background to the concept of purge factor calculation and introduces a consortium-led initiative, the Lhasa Mirabilis Project.4 The Mirabilis consortium seeks to develop an industry-standardised approach, providing an expert and scientifically robust software for the automated calculation of purge factors for potentially mutagenic impurities in a synthetic route.
Tove Tuntland
Genomics Institute of Novartis Research Foundation (GNF), USA
Title: Target Based vs. Phenotypic Screening Approaches in Drug Discovery

Biography:
Tove Tuntland holds a Pharmacy degree from the University in Oslo, Norway, and a Ph.D. in Pharmaceutics from the University of Washington, Seattle, USA. She has expertise in preclinical drug metabolism and pharmacokinetics (DMPK), pharmacology and PK/PD, and worked in discovery and development at Pfizer Global Research and Development (PGRD) in La Jolla, California (1996 to 2002). Thereafter until present time she has led a group at Genomics Institute of Novartis Research Foundation (GNF), supporting in vitro and in vivo preclinical DMPK and PK/PD studies in a variety of discovery and development programs in oncology, immunology, infectious and metabolic diseases.
Abstract:
Target-based drug discovery can effectively develop novel treatments for a validated target, but the process of target validation is complex and associated with high degree of uncertainty[1]. As an alternative to the target based approach, phenotypic screening is making a comeback in drug discovery. Such assays characterize phenotypic events related to disease modification and do not require prior understanding of the mechanism of action[2]. At GNF/Novartis, scientists have used both target based and phenotypic screening approaches to successfully identify novel drugs. Target based methods lead to the discovery of ceritinib (Zykadia™, formerly LDK378), a highly potent and selective anaplastic lymphoma kinase (ALK) inhibitor which was recently approved by the FDA for the treatment of patients with ALK-positive metastatic non-small cell lung cancer (NSCLC) who were previously treated with crizotinib. Phenotypic screening led to the discovery of cipargamin (KAE609), the first new antimalarial drug candidate with a completely novel mechanism of action to reach phase 2 clinical development in over 20 years. When tested clinically in adults with uncomplicated P. vivax or P. falciparum malaria, cipargamin was shown to clear parasitemia after 3 days of repeated dosing. Also identified by phenotypic screening, the novel antimalarial drug KAF156 demonstrated activity against both liver and blood stage malaria, including artemisinin-resistant parasites. The approaches used to discover and develop the novel drugs ceritinib, cipargamin and KAF156 will be discussed.
Sergey Lednev
P.G. Demidov Yaroslavl State University, Russian Federation
Title: Analysis of the polar solvents influence on the mechanism of the inhibited oxidation of unsaturated compounds

Biography:
Sergey Lednev has completed his PhD at the age of 27 years from P.G. Demidov Yaroslavl State University. He is an assistance lecturer at the Department of General and Physical Chemistry. He has published 2 papers in reputed journals.
Abstract:
Polar solvents are widely used in the pharmaceutical industry and in various fields of applied chemistry. The ability to dissolve many organic substances and the wide temperature limits of the liquid state make polar solvents essential components of reaction mixtures in industrial and biotechnological processes as well as the components of drugs. On the other hand, polar solvents determine the medium in which the chemical process proceeds, and often have a significant effect on its kinetics. Therefore, an important practical task is to take into account the influence of the medium polarity on the kinetics of chemical reactions.
Thus, the medium polarity can have a significant effect on the reactivity of unsaturated compounds during the oxidation by molecular oxygen. The study of this process is important both for chemical technology and for understanding the chemistry and biology of oxidative stress. The effects of nonspecific and specific solvation may have a significant effect on the mechanism of oxidation of unsaturated compounds in the medium of polar solvents. The addition of an oxidation inhibitor to the reaction mixture complicates these effects. The report discusses the results of a systematic study of specific and nonspecific solvation effect on elementary stages of the inhibited oxidation of unsaturated compounds (methyllinoleate, styrene, methyl methacrylate, butyl acrylate). Antioxidants are phenols (PhOH), aromatic amines (AmH), stable nitroxide radicals (>NO•), and corresponding hydroxylamines (>NOH). The results were obtained using a modern analytical basis: high-sensitivity microvolume, FT-IR, and NMR spectroscopy.
The research is supported by RSF grant No. 14-23-00018.
Victor Shtamburg
Ukrainian State University of Chemical Technology, Ukraine
Title: Arylglyoxals as precursors for synthesis of N-hydroxyhydantoins, N-alkoxyhydantoins and thiohydantoins in mild conditions

Biography:
Victor Shtamburg has completed his PhD at the age of 26 years from Dnipropetrovsk National University (Ukraine). He is chemist and scientist of Ukrainian State University of Chemical Technology (Dnepr). He has published more than 14 papers in reputed journals.
Abstract:
It is known that hydantoins are used as drugs and precursors for drugs synthesis. But the easy method for synthesis of N-hydroxyhydantoins and N-alkoxyhydantoins was not known before. We have proposed that interaction of arylglyoxals with N-hydroxyurea or with N-alkoxyureas may be a simple route to 3-hydroxy-5-arylhydantoins and to 3-alkoxy-5-arylhydantoins respectively. As we found, arylglyoxals reacted with N-hydroxyurea in an aqueous solution at room temperature according to exact scheme. At the first stage the substituted urea formed. At the second stage urea cyclizes into 3,4,5-trihydroxyimidazolidine-2-ones. Compounds were protonized by N-hydroxyurea with further elimination of the water molecule from С-5 atom. Then 1,2-shift of hydrogen atom from atom С(4) to atom С(5) occurs, yielding 3-hydroxyl-5-arylhydantoin. This interaction may be stopped on different stages. It depends not only on nature of arylglyoxal, but also on the reaction conditions. In these conditions the products of stages I, II, III may be isolated depending of the arylglyoxal structure and temperature. Thus, the reaction of arylglyoxals with N-hydroxyurea in acetic acid at room temperature selectively yields only 3-hydroxy-5-arylimidazolidine-2,4-diones. The reaction of arylglyoxals with N-alkoxyureas in acetic acid at room temperature also selectively yields 3-alkoxy-5-arylimidazolidine-2,4-diones. Also in acetic acid arylglyoxals react with thiourea selectively yielding 5-arylimidazolidine-4-one-2-thiones at room temperature.

Biography:
Feng ZHANG has completed his PhD at the age of 26 years from Second Military Medical University, and has served as an assistant Professor in Clinical Pharmacy for Drug Safety and Effectiveness since her graduation. For chemical drugs, she promotes better standardization of analytical practices in LC-MS based therapeutic drug monitoring analyses in clinical application. She has published more than 11 SCI papers as (co)first author, and is responsible for four funds as a project leader. She also has applied for 4 Chinese patents.
Abstract:
Multiple myeloma (MM), a malignant neoplastic serum-cell disorder, has been a serious threat to human health. Determination of 6 commonly used drug concentrations, including thalidomide (THD), lenalidomide (LND), cyclophosphamide (CTX), bortezomib (BTZ), dexamethasone (DXM) and adriamycin (ADM), in MM patients was of great clinical interest. Drug pharmacokinetic monitoring was important to ensure safety and efficacy during chemotherapy. We reported a method for the rapid and simultaneous measurement of the above therapeutics by liquid chromatography-tandem mass spectroscopy (LC–MS/MS) method with solid phase extraction. Analysis was performed on a Waters XBridge®BEH C18 column (2.5 μm, 2.1mm × 50 mm), with formic acid aqueous solution and acetonitrile as the mobile phase at flow rate 0.3 mL/min. All analytes showed good correlation coefficients (r > 0.996), and LLOQ of THD, LND, CTX, BTZ, DXM and ADM were 4, 2, 2, 2, 2 and 2 ng/mL, respectively. The inter- and intra-day precisions and stability were expressed as variation coefficients within 15% and relative error less than 15%. Dilution effect, carryover and incurred sample reanalysis were in the acceptable levels. Method validation was investigated according to the 2015 edition Chinese Pharmacopoeia guidelines, as US FDA (2013, revision 1) required. The descried method was successfully applied to determine the clinical incurred serum samples from MM patients, and the tested results were also provided to the doctors for consideration. The LC–MS/MS based assay may improve future clinical studies evaluating common therapeutics for MM treatment.
Venkatesan Jayaprakash
Birla Institute of Technology, India
Title: Designing antitubercular agents exploring Mycobactin biosynthetic patway

Biography:
Dr. Venkatesan J. has completed his PhD from Birla Institute of Technology, Mesra in the year 2012. He is currently serving as Associate Professor in Department of Pharmaceutical Sciences & Technology, BIT, Mesra. He has around 50 publications to his credit in the area of drug design and medicinal chemistry. His research group in BIT is currently working in the area of drug design against the validated targets of tuberculosis and emerging virus like Dengue and Chikungunya. He is also serving as Editor of Journal of Pharmaceutical Chemistry.
Abstract:
Developing newer antitubercular agents having novel chemical scaffold targeting novel proteins is essential to face the threat due to MDR and XDR tuberculosis. Mycobactin is a hexadentate ligand secreted by the tubercular bacilli to overcome the iron stress that it experiences once inside the host cell. Inhibiting any enzyme in the biosynthetic pathway will be novel approach in developing a newer chemotherapeutic agent against tuberculosis. Our group is working on mimics of phenyloxazoline portion of mycobactin. We identified, 3-(2-hydroxyphenyl)-5-(4-hydroxyphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (1) as a potential inhibitor of Mycobacterium tuberculosis under iron stressed condition (Bioorg Med Chem Lett, 2008, 18(8):2662-8). We hypothesize that this compound as a putative inhibitor of phenyloxazoline synthetase, an enzyme in the mycobactin biosynthetic pathway catalysing the condensation/cyclization salicylic acid and serine. Compound 1 is a racemic mixture and their component isomers were found to be equipotent in nature (unpublished data). Further modification at the 1N of pyrazoline replacing the –C(=S)-NH2 group with –C(=O)-NH2, –C(=NH)-NH2 and –C(=O)-CH3 resulted in the reduction/loss of activity (unpublished data). With this background, analogue of compound 1 with different substitutions on the phenyl ring at 5th position of pyrazoline were designed, synthesized and evaluated for the antitubercular activity under iron stress using Mycobacterium smegmatis. Compounds were also subjected to in vitro cytotoxicity studies. Potent compound from this series is currently under screening against Mycobacterium tuberculosis and we are also planning for similar studies against resistant strains of Mycobacterium tuberculosis.
Mustapha A. Tijjani
University of Maiduguri, Nigeria
Title: Isolation and Structural Elucidation of 20 hydroxyecdystone from Vitex doniana Sweet Stem bark (Black plum)

Biography:
Abstract:
Vitex doniana Sweet, a plant commonly known black plum, in English, Prunier noir in French, dinya in Hausa , ucha koro in Igbo, oori-nla in yoruba and ngarmi in Kanuri is a medium-sized deciduous tree, 8-18m high, with a heavy rounded crown and a clear bole up to 5m. V. doniana is from Verbenaceae family and abundantly occurring in savannah regions. It can be found throughout tropical Africa.The ethanolic extract of Vitex doniana stem bark (11.9g) was subjected to a silica gel accelerated column chromatography and eluents fractions (150ml aliquots) obtained were collected and monitored with thin layer chromatography (TLC).. Fractions with similar Rf values from same solvents system were poled together. Phytochemical test of all the fractions were perform. Complete elution yielded 48 fractions (150ml/fraction) which were pooled to 24 fractions and finally to eight (8) eight fractions and coded. Fraction Vd8-a (56mg) has gave a single spot a white crystal compound coded V1 on checking with TLC and observed under Ultraviolet lamp .The Rf values was calculated to be 0.433 and melting point was found to be 241-243°C uncorrected. The infra red spectrum of compound V1 shows prominent peaks that corresponds to OHstr (3365cm-1) and C=0 (1652cm-1). The 1H NMR (400 MHZ) spectrum of compound V1 in DMSO-d6 displayed five singlet signals. It further showed a broad singlet at δ 5.58 integrated for 1 H is due to an olefinic H-atom adjacent to the carbonyl carbon atom. Three signals at δ 3.10` (d, J = 9.0 Hz, H-22), 3.59 (m, 1H, 2H-a) and 3.72 (m, 1H, 3H-e) each integrating for one proton is due to an oxymethine protons indicating that three oxymethine H-atoms were present in the compound. The 13C-NMR spectrum showed the presence of 27 Carbon atoms, suggesting that may be steroid skeleton and The DEPT-135 spectra showed the presence of five CH3, eight CH2, and seven CH groups, and seven quaternary C-atoms. The Molecular formula was established as C27H44O7 by HRES-MS positive ion mode m/z 481.3179. Based on the spectral analysis, the compound V1 is thus concluded to have ecdysteriod skeleton and conclusively conforms with 2β, 3β 14α, 20R, 22R, 25- hexahydroxy-5 β cholest-7-ene-6- one, commonly known as 20-hydroxyecdysone. This is the first time this compound was isolated from vitex doniana sweet.
Gervais Bérubé
Université du Québec à Trois-Rivières, Canada
Title: From para-aminobenzoic acid to small compounds with multiple biological properties

Biography:
Gervais Bérubé completed his Ph.D. in 1986 in the field of Organic Chemistry at the Université de Sherbrooke. After completing three post-doctoral stays initially in the field of Organic Chemistry, then in Oncology and lastly in Immunology, he became assistant professor of Medicinal Chemistry in the School of Pharmacy, Memorial University of Newfoundland. He is now professor of Organic Chemistry at the Université du Québec à Trois-Rivières. His main research interest is the design and development of new anticancer drugs. He has published more than 76 papers in reputed journals and presented more than 110 communications in diverse meetings.
Abstract:
The discovery of new molecules possessing multiple biological properties in a single entity is a subject of great interest to the scientific community. A simple compound previously used in our laboratory as a heterobifunctional linker to construct immunoconjugates was identified as a potential lead drug showing anti-inflammatory, anti-metastatic as well as anticancer activities. The lead compound is made in only three chemical steps from para-aminobenzoic acid with 43% overall yield. Its structure can be modified to give alternative analogs with similar activities. The prospect of lead optimization is significant. This communication will highlight the chemical and biological potential (in vitro and in vivo) of these type of molecules tested on bladder cancer. Amongst the results, the lead compound can reduce the size of a tumor in an animal model by 90% within 25 days without apparent side effects.
Mohan Paul Singh Ishar
Maharaja Ranjit Singh Punjab Technical University, India
Title: Trysts with Anti-Cancer Drug Designing – Design Synthesis and Evaluation Some Chromone and β-Ionone Based Anticancer Molecules

Biography:
Prof. Mohan Paul Singh Ishar, an alumnus of Indian Institute of Technology, Delhi, is presently serving as Vice Chancellor, Maharaja Ranjit Singh Punjab Technical University, Bathinda (Punjab) India. With more than 1500 citations, 120 papers in reputed journals and 70 presentations in conferences, he is leading a research team focusing on Synthetic Organic and Medical Chemistry; the Design Synthesis and Evaluation of Biologically Active Compounds, Anti-Cancer Drugs and Anti-Microbial Agents, Allene Chemistry, Steroid Chemistry and Organic Photo Chemistry. and also serving as an editorial board member of repute.
Abstract:
The systematic discovery of new drugs, also called “Drug Designing”, involves envisioning, preparation and systematic evaluation of specific new molecules leading to more efficient drug discovery. A modern drug designing research is a frontier area which requires inputs from diverse disciplines such as Natural product chemistry, Synthetic chemistry, Computational chemistry, Spectroscopic techniques- in particular NMR and X-ray crystallography, Biochemistry, Physiology, Genomic technologies, Bioinformatics, Molecular biology, Microbiology, Pharmacology, etc. A number of approaches are being adopted for discovery of new “lead” structures. Despite tremendous advancements made in synthetic methodologies, natural products continue to be the most consistent source of new exotic molecular frameworks fore drug discovery. Despite tremendous advancements in identification of new targets for chemotherapeutic intervention in fight against cancer and combination regimens of available anti-cancer drugs, problem of adverse effects and developing resistance of cancer cells to drugs have made many chemotherapeutic regimens ineffective. Therefore, the search for novel targets for anticancer drugs and more effective chemotherapeutic agents for the treatment of cancer is highly desired. The presentation enlists modern targets for anticancer drug designing program and includes examples form our own work on design, synthesis and systematic evaluation of some chromone and β-ionone based cytotoxic agents obtained through environmentally benign synthetic protocols.
Roberta Ettari
University of Messina, Italy
Title: Development of inhibitors of the cysteine proteases rhodesain of T. b. rhodesiense and falcipain-2 of P. falciparum

Biography:
Roberta Ettari in 2008 received her Ph.D. in Pharmaceutical Sciences at the University of Messina, she then worked as a postdoctoral researcher at the School of Chemistry (Galway, Ireland) and at Messina and Milan Universities. She spent also some research periods as guest scientist at Merck Research Laboratories (Pomezia), Instituto Grifols (Barcelona), Würzburg and Mainz Universities (Germany). In 2011, she received the “Farmindustria National Award for the Excellence in Medicinal Chemistry”. Since 2014, she is Assistant Professor of Medicinal Chemistry at the University of Messina working on the development of inhibitors of proteases involved in human and parasitic diseases.
Abstract:
Neglected tropical diseases (NTDs) are a group of disabling infections particularly endemic in developing regions of Africa, Asia and the Americas. Over one billion people suffer from one or more NTDs, two of the most important are Human African Trypanosomiasis (HAT) and malaria. Although a number of antitrypanosomal and antimalarial agents are available, these suffer from problems like increasing drug resistance, toxicity and route of administration. Thus, there is an urgent need to identify new effective drugs, ideally directed against novel targets. The cathepsin L-like cysteine proteases rhodesain and falcipain-2 (FP-2), have been recognized as novel promising targets for the treatment of HAT and malaria respectively, because of their key roles for parasite survival.
The importance of rhodesain, a cysteine protease of T. brucei, is due to its several functions, such as its role in crossing the blood brain barrier, thus inducing the neurological stage of HAT; other functions include the turnover of variant surface glycoproteins that coat trypanosomes, degradation of host immunoglobulins to reduce the host immune response, and degradation of parasite and imported host proteins within lysosomes. On the other hand, FP-2, the main cysteine protease of P. falciparum, hydrolyzes hemoglobin to provide amino acids that are essential to the parasite for protein synthesis. FP-2 may also be responsible for the cleavage of the cytoskeletal proteins ankyrin and band-4.1 to facilitate rupture of the red-cell membrane. Thus, the development of novel rhodesain and FP-2 inhibitors is a promising challenge to obtain new effective agents for the treatment of HAT and malaria.
Evgeny Pliss
P.G. Demidov Yaroslavl State University, Russian Federation
Title: Oxidative stress simulation by radical-chain oxidation of vinyl compounds

Biography:
Evgeny Pliss has completed his Candidate of Science degree in 1978 and Doctor of Science in 1990. He has published more than 90 papers in reputed journals and is an author of several monographs. He is chair of General and Physical Chemistry at P.G. Demidov Yaroslavl State University.
Abstract:
A combination of microvolumetry, the rotating sector method, ESR, 1H-NMR, and IR allowed to establish a detailed mechanism of liquid-phase oxidation of vinyl compounds. A distinctive feature of the mechanism is the fact that the oxidation chain is carried out by a low-molecular hydroperoxide radical. Kinetic, correlation, and quantum-chemical analysis of these processes allows modeling the processes leading to oxidative stress in living organisms. The report analyses the routes of various chemical reactions that reduce the negative impact of oxidative stress. The study is proposes the use of a stable nitroxyl radicals of piperidine, pyrroline and imidazoline series as one of the effective components, leading to a positive effect.
The research is supported by RSF grant No. 14-23-00018.
Milan Mladenović
University of Kragujevac, Rebublic of Serbia
Title: Monoamine Oxidase B inhibitors as Leads in Parkinson’s Desease Treatment: Rational Design of Novel Scafolds Driven by Structure-Based 3-D QSAR

Biography:
Milan Mladenović has completed his PhD at the age of 27 years from University of Kragujevac, Republic of Serbia and postdoctoral studies the age of 27 from Sapienza Universiry of Rome. He is an Assistant Professor of Chemistry of Natural Products and Biochemistry at Faculty of Science, University of Kragujevac, and a Head of Department of Biochemistry. He has published more than 25 papers in reputed journals in the field of Biochemitry and Medicinal Chemistry. In the year of 2013 he became a director of Kragujevac Center for Computational Biochemistry, a specialized laboratory for molecular modeling and drug design.
Abstract:
Monoamine oxidase B (MAO B) catalyzes the oxidative deamination of aryalkylamines neurotransmitters with concomitant reduction of oxygen to hydrogen peroxide. Consequently, the enzyme’s malfunction induces oxidative damage to mitochondrial DNA and mediates development of Parkinson’s disease. Aiming to develope the novel reversible MAO B inhibitors, novel 128 compounds were rationaly designed after the modification of higly active co-crystallized coumarin inhibitor (Protein Data Bank ID: 2V61), utilizing the structure-based (SB) 3-D QSAR/SB and ligand-based (LB) alignment assesment/in vitro evaluation protocol. Coumarin 2V61 was improved by rules defined by 3-D QSAR models, generated from the available co-crystallized inhibitor-MAO B complexes, utilizing the 3-D QSAutogrid/R procedure: (1) an isobutyramide, as hydrogen-bond donnor towards MAO B Glu206, was introduced to the coumarin C3 position; (2) the C4 carbon was saturated with mixed hydrogen-bond acceptor/hydrophobic functions to interfere with recongition cage residues Tyr398 and Tyr435; (3) the C3=C4 double bond was improved with 1-methyl-6-oxopiperidine-2-carboxamide or 2-oxo-1,2,3,4-tetrahydropyrimidine-4-carboxamide rings and their methylated forms; (4) the C5 and C6 carbons were substituted by hydroxyl/methoxy functions to interact with Cys172; (5) the m-chlorobenzyloxyl moiety at C7, upgraded with o-, m-, or p-methyl groups, was retained to force the Ile199 in open conformation and to activate Phe103. Designed structures were SB LB aligned against MAO B, and six compounds were experimentally validated through synthesis and biological evaluation in vitro. Of them, two compounds, D123 (IC50 = 0.83 nM, Ki =0.25 nM) and D124 (IC50 = 0.97 nM, Ki =0.29 nM) were distinguished as potential lead candidates as anti-Parkinson’s drugs.
Matthew D. Lloyd
University of Bath, UK
Title: High-Throughput Screening to identify novel inhibitors of human α-methylacyl-CoA racemase 1A (AMACR; P504S)

Biography:
Matthew D. Lloyd graduated with a DPhil from Oxford on clavulanic acid biosynthesis. Following post-doctoral research at Brown University, U.S.A. and Oxford, he took up a lectureship at the University of Bath in 2002. He is currently Senior Lecturer (Associate Professor) in Pharmacy & Pharmacology. Research interests include prostate cancer, chemical biology, lipid metabolism, enzymes and inhibitors as drugs. He has published >70 research papers and is an editorial board member of The Journal Of Enzyme Inhibition and Medicinal Chemistry and The World Journal of Biological Chemistry. He is also a 4th Black Belt (International Instructor) in the Ch’ang-Hon Taekwon-Do.
Abstract:
α-Methylacyl-CoA racemase (AMACR; P504S) catalyzes a key step in the degradation of branched-chain fatty acids and is important for the pharmacological activation of Ibuprofen and related drugs. Both the concentration and activity of AMACR are increased in prostate and other cancer cells, and the enzyme is a recognized drug target. However, all of the reported inhibitors are acyl-CoA esters (which do not comply with Lipinski guidelines) or non-specific protein modifying agents. Libraries of ~20,000 drug-like compounds were screened using a novel colorimetric assay; Incubation of R,S-2-3-(2,4-dinitrophenoxy)-2-methylpropanoyl-CoA with active AMACR resulted in the elimination of the strongly yellow 2,4-dinitrophenoxide and allows continuous measurement of activity in a microtitre plate format. Inhibitors were identified by a reduction in the rate of reaction in the presence of the library compound vs. the control. A number of novel reversible inhibitors were identified and their potency determined using dose-response curves. The results demonstrate the utility of the assay for the discovery and characterization of AMACR inhibitors as anti-cancer agents.
This work was funded by Prostate Cancer UK (PG14-009), a Biochemical Society Summer Vacation Studentship Award, The Nuffield Foundation and MRC technology.

Biography:
Shouhong GAO has completed her Master degree of Pharmacy at the age of 27 year in Second Military Medical Univerisity. She has served as an associate Professor in Clinical Pharmacy for Drug Safety and Effectiveness since her graduation. For chemical drugs, she promotes better standardization of analytical practices in LC-MS based therapeutic drug monitoring analyses in clinical application. She has published more than 9 SCI papers as (co)first author, and was responsible for five funds as a project leader.
Abstract:
First-line anti-tuberculosis drugs are playing vital roles for curbing rapid spread of tuberculosis. mutidrug therapies are commonly applied in clinical to achieve better treatment outcomes. However, drug resistance and adverse reactions came along with this therapies and therapeutic drug monitoring is a feasible way to precaution side effects. For this reasons, a simple and sensitive method based on LC-MS/MS and single protein precipitation was developed and validated for simultaneously quantifying of pyrazinamide, isoniazid, ethambutol, streptomycin and rifampicin in human plasma. Optimized chromatographic separation was achieved on a ZORBAX SB-C18 column with heptafluorobutyric acid, an ion-pair reagent, in the mobile phase at a flow rate of 0.3mL/min. The mass detection was achieved using electrospray ionization in the positive ion mode with a multiple reaction monitoring scan: m/z 124.1→79.1 for pyrazinamide, m/z 138.1→120.9 for isoniazid, m/z 205.3→116.2 for ethambutol, m/z 582.3→262.6 for streptomycin, m/z 823.4→791.2 for rifampicin and m/z 180.1→110.1 for phenacetin(Internal standard, IS). The LLOQ of pyrazinamide, isoniazid, ethambutol, streptomycin and rifampicin was 200, 80, 0.2, 2000, 200ng/mL, respectively. The Intra-day and inter-day accuracy and precision were within 15.0%. The method had been successfully applied to simultaneous determination of four first-line Anti-tuberculosis drugs in plasma from tuberculosis patients.
Geronikaki Athina
Aristotle University of Thessaloniki, Greece
Title: Study of inhibition of the protein phosphatases ptp1b, lar and hsp-2 by novel 3-((furan-2-yl)methyl)-2-phenyl thiazolidin-4-one derivatives. potent drugs for the treatment of diabetes and obesity

Biography:
Abstract:
Phosphorylation-dephosphorylation is a predominant mechanism involved in the regulation of eukaryotic cell function. During the last decade, protein kinases and phosphatases have been extensively studied and members of both enzyme families have been proved to be related with various diseases. As a result drug research has been focused in specific kinase or phosphatase regulation. PTP1B is a protein tyrosine phosphatase, acting on insulin receptor. Interaction of insulin with its receptor results in changes in the intracellular structure of the receptor followed by the phosphorylation of three tyrosine residues. This is the first step of a cascade of intracellular events leading to glucose uptake. The cascade is terminated by the dephosphorylation of insulin receptor by PTP1B. Thus, PTP1B inhibition, resulting in prolonged maintenance of the phosphorylated state, practically enhances insulin effect. Consequently, effective and selective PTP1B inhibitors can be potent drugs for the treatment of type 2 diabetes and obesity. LAR, a receptor-like transmembrane PTP is also believed to be a target for insulin receptor signalling, as LAR inhibitors were found to dramatically potentiate insulin's effects. Moreover, it was found that it is concentrated in mature synapses in hippocampal neurons, and is believed to play a role in the development and maintenance of excitatory synapses. HSP-2 is one of the classical non-transmembrane PTPs. A mutation in the gene, making HSP-2 continuously active results in Noonan syndrome, a developmental disorder affecting 1 in 2500 newborns that may lead to a higher risk for certain cancers, including juvenile myelomonocytic leukaemia. Treatment with HSP-2 selective inhibitors could bring hope to Noonan Syndrom patients. Although, many compounds have been tested as PTP1B inhibitors, the race for finding a highly effective and absolutely selective inhibitor continues. Most inhibitors act on more than one protein phosphatases. In the present study, we synthesised a number of 3-((furan-2-yl)methyl)-2-phenyl thiazolidin-4-one derivatives and tested their PTP1B inhibitory activity as well as the structure – activity relationship. Many of the derivatives showed good inhibitory action. Moreover, we evaluated the specificity of the compounds by testing the inhibition of protein phosphatases LAR and SHP-2. All inhibitory actions were tested using human recombinant phosphatases. In all cases p-nitro-phenyl-phosphate was used as substrate.

Biography:
Abstract:
The present work describes the preparation of protected octapeptide derivative (23-30) B-cain human insulin and coupling this peptide to B-chain desoctapeptide iodinated at tyrosine B16. B-chain will recombine with natural A-chain to give human insulin iodinated at tyrosine B16. Our approach of synthesis consisted of using stepwise coupling fragment condensation. All coupling was successfully with a high yield and easy to purify. For the side chain protection, we used (But) for OH-group of threonine and tyrosine, and (But) ester for C-terminal threonine. In most cases, Carbobenzoxy group was used for protection of N-terminal amino acids (Lys,Thr,Gly). Methylsulphonylethyloxycarbonyl (MSC) group was used for protection of NS-lysine.
Hattab Youcef
University of Science and Technology of Oran M.B., Algeria.
Title: Synthesis and characterizations of organophile MMT –copolymer (St-THF) nanocomposite

Biography:
Abstract:
Nanocomposites, are mixed materials synthesized from one or two polymers blended in optimized percentages and Organophilic clay. The objective of this study is to use the Organophilic clay (Maghnite: montmorillonite) in the presence of tow monomers to obtain a copolytetrahydrofuran- styrene nanocomposite by in-situ polymerization. Organophilic montmorillonite (O-MMT) was prepared by ion exchange between Na+ ions in the clay hexadecyltrimethylammonium bromine cations in an aqueous medium. The organophilic MMT particles were easily dispersed in styrene (St) and tetrahydrofuran (THF) monomer. The structure of obtained modifier was investigated by proton nuclear magnetic resonance (RMN1H), Fourier-transform infrared (FT-IR) spectroscopy. The exfoliating structure of nanocomposite was probed by X-ray diffraction (XRD) and transmission electron microscopy (TEM). The Thermal stability were also investigated with thermogravimetric analyzer (TGA), comparing with pure polystyrene, the nanocomposite showed much higher decomposition temperature.
Prakash Kinthada
Sri Vidyanikethan Engineering college, India
Title: Transition metal complexes/organometallic compounds as anticancer/anti HIV drugs or in pharmaceutical industry

Biography:
Prakash Kinthada is a Professor in Chemistry at Sri Vidyanikethan Engineering college, JNTU University in Ananthapur, A. Rangam Peta, Tirupathi, India.
Abstract:
Cancer is a dreadful disease and any practical solution in combating this disease is of paramount importance to public health. Cancer patients have burdened by drug induced toxic side effects, and no turned to seek help from the complementary and alternative medicine hoping for a better cure. Research on Platinum based drugs and Non Platinum based drugs is a Multi-Million Dollar Industry in USA and there is every need to produce safe drugs for the cure of this monstrous disease. Flavonoids have a long history of use in traditional medicines in many cultures. The phytochemical, curcumin is one of the major dietary flavonoid, belonging to a group of flavonol, Curcumin is a natural polyphenol. It is highly potential molecule capable of preventing and treating various cancers. Various dietary chemo preventive agents, turmeric powder or its extract are broadly used as therapeutic preparations in Indian System of medicine. We provide a summarized synthesis and structural determination of Curcumin Oxime, Curcumin Thiosemicarbazone derivative of Gold (III) complex. The use of these analogs for prevention of cancer tumor progression and treatments of human malignancies. A pharmacologic agent for treating and/or preventing cancer, among other diseases and conditions, and particularly breast, prostate, and pancreatic cancer, in humans and animals. The novel pharmacologic agent is an isoflavonoid or isoflavonoid mimetic covalently attached to a cytotoxic pharmacophore that, preferably has the ability to conjugate with a metal salt to form a more potent metal complex, particularly a Au (III) complex and other complexes of Platinum, Palladium, Ruthenium, Copper etc. My talk would mainly encompass different Transition Metal Complexes/Organometallic Compounds that are presently used as drugs, especially Anticancer and Anti-HIV drugs, apart from Anti-inflammatory, Antimicrobial, Antibacterial and diseases like Arthritis and Parkinson’s Disease etc. The talk would mainly focus on the use of Medicinal Chemistry and it’s application to Drug Design and Development in Pharmaceutical Industry , especially Transition Metal Complexes and Organometallic Compounds viz. Gold, Platinum, Palladium And Ruthenium apart from Copper, Cobalt, Iron, Nickel, Zinc, Cadmium etc. The main emphasis of my talk would be on Different class of Ligands, their Schiff’s Bases and Transition Metal Complexes especially Au, Pt, Pd and Ru, with the main aim of designing, developing very novel small molecules, as possible and extremely potential candidates as Anti-cancer and Anti-HIV drugs. The talk would provide an overview of current programs being undertaken in our laboratories, especially focused on the development of potent ligands capable of recognizing Binding sites and diverse strategies employed by my group for elucidation of Anti-Cancer and Anti-HIV drug Leads to Circumvent the problem caused by Cis-Platin. We have synthesized and characterized several phytochemicals from Traditional Medicinal Plants and isolated some phytochemicals and made the corresponding Oximes, Thiosemicarbazones and Substituted thiosemicarbazones as ligands and synthesized, characterized, structurally elucidated their Transition Metal Complexes especially with Gold, Platinum, Palladium, Ruthenium, Copper etc. and Studied their Anticancer Activity, Nuclease activity etc. and tested their potential as Anticancer Drugs. The main aim of our extensive/preclinical Pharmaceutical development program is to investigate the use of these extremely novel small molecules-metal complexes/compounds of phytochemicals, flavanoids etc., which have very interesting structural features and properties and hence are excellent candidates as Anti-Cancer and Anti-HIV drugs .The main aim of our research is Design ,Development and Synthesis of Transition Metal Complexes/ Organometallic Compounds that would certainly help to bring this force of nature from BENCH to BEDSIDE and enhance Cancer Killing with less toxic effects and would certainly lead to initiation of clinical trials.

Biography:
Tetsuo Narumi has completed his Ph.D at the age of 28 years from Kyoto University with Prof. Nobutaka Fujii. He spent a year in US as a JSPS postdoctoral fellow with Prof. Jeffrey W. Bode at the University of Pennsylvania. In 2009, he began his academic career in Japan, at Tokyo Medical and Dental University with Prof. Hirokazu Tamamura. In 2013, he began his independent career at Shizuoka University, in Japan, as an associate professor in the Bioorganic Chemistry. He has published more than 50 papers in reputed journals and serving as a leading researcher in the fields of peptidomimetic science.
Abstract:
N-Heterocyclic carbenes (NHCs) have become widely utilized as organocatalysts in the formation of challenging C-C bonds. In particular, conjugated Breslow intermediates, derived from the reaction of a,b-enal and NHCs, have emerged as a powerful tool for a variety of transformations. Since the concept of conjugated umpolung through the intermediacy of catalytically generated homoenolates and activated carboxylates, many investigations have revealed their utility to synthesize various heterocycles such as γ-butyrolactones, γ-lactams and cyclopentenes. However, there are few methods for tuning the catalytic reactivity of NHCs by either rational design of NHC catalysts or appropriate additives for cooperative catalysis. This is particularly true of imidazolylidene catalysts, only a few members of which are utilized effectively for NHC catalysis. In this study, structural and kinetic investigation of a series of imidazolyli-dene catalysts with various N-aryl groups for homoenolate-mediated g-butyrolactone formation. Our study revealed that the reaction rate and catalytic reactivity can be clearly increased by the combination of 2,6-diethylphenyl groups as N-aromatic rings and a small excess amount of DBU as base compared to the originally reported conditions. Details of the structural studies, mechanistic investigations including parallel KIE experiments, and applications to the design of novel imidazolylidene catalysts will be discussed.
Sabu Thomas
Mahatma Gandhi University, India
Title: Engineering at the Nanoscale: A Strategy for Developing High Performance Functional Materials for Tissue Engineering

Biography:
Professor Sabu Thomas is presently the Director and Professor of International and Interuniversity Centre for Nanoscience and Nanotechnology and School of Chemical Sciences, Mahatma Gandhi University, India. He has more than 30 years’ experience in Polymer Science and Technology and has contributed greatly to the research and development of Nanoscience and Nanotechnology. Prof. Sabu Thomas received his Ph. D from IIT, Kharagpur and then joined as a Senior Visiting Researcher, Katholieke University, Leuven, Belgium and Laval University, Quebec, Canada. He is one of the pioneers of polymer science and technology and has published over 750 peer reviewed research papers.He has co-edited nearly 60 books and is the inventor of 5 patents. He has supervised 79 Ph.D theses and his H index is 79 with nearly 29,675 citations. Prof. Thomas has delivered over 300 Plenary/Inaugural and Invited lectures in national/international meetings over 30 countries. He is currently, the chief editor of Nano-Structures & Nano-Objects and also serves the reviewer for several International journals. Professor Thomas has received a number of national and international awards including, FRSC,MRSI, Nano Tech, CRSI, Sukumar Maithy Award etc. He is in the list of most productive researchers in India and holds a position of No.5. Thomas has been conferred Honoris Causa (D.Sc) by the University of South Brittany, France in 2015 and in 2017 he has been selected for conferring with D.Sc from University Lorraine, France.
Abstract:
The talk will concentrate on various approaches being used to engineer materials at the nanoscale for various applications in future technologies. In particular, the case of clay, carbon nanostructures (e.g. nanotubes, graphene), metal oxides, bionanomaterials (cellulose, starch and chitin) will be used to highlight the challenges and progress. Several polymer systems will be considered such as rubbers, thermoplastics, thermosets and their blends for the fabrication of functional polymer nanocomposites. The interfacial activity of nanomaterials in compatibilising binary polymer blends will also be discussed. Various self assembled architectures of hybrid nanostructures can be made using relatively simple processes. Some of these structures offer excellent opportunity to probe novel nanoscale behavior and can impart unusual macroscopic end properties. I will talk about various applications of these materials, taking into account their multifunctional properties. Some of the promising applications of clay, metal oxides, nano cellulose, chitin, carbon nanomaterials and their hybrids will be reviewed. Finally the effect of dewetting up on solvent rinsing on nano scale thin films will also be discussed.
Larisa.Klapshina
Nizhny Novgorod State University/IOMC RAS, Russia
Title: Novel sensitive sensors of intracellular viscosity and potential anticancer theranostic agents prepared on the porphyrazine pigments platform for specifically personalized medicine.

Biography:
Larisa Klapshina received her Ph. D in 1992 from the Razuvaev Institute of Organometallic Chemistry of Russian Academy of Sciences, IOMC RAS, (Nizhny Novgorod, Russia). Currently she is senior researcher at IOMC RAS and at the Laboratory of Optical Theranostics in Nizhny Novgorod State University). She and her group work in the area of organic and organometallic synthesis and the functional materials for biophotonics and biomedicine. She is author of about 100 artcle
Abstract:
The various techniques available for cancer diagnosis and therapy are traditionally considered as separate approaches in medical care. But nowadays development of the multifunctional agents which combine modalities for cancer diagnosis, treatment and real-time monitoring of treatment progress is real imperative for specifically personalized medicine. Here we report series of novel tetracyanotetra(aryl) porphyrasine dyes which are found to be red-emitting fluorescent ‘molecular rotor’ i.e. the fluorescence lifetime and the quantum yield of these macrocycles strongly increase as a function of environment viscosity. On the other hand, they works as an efficient PDT sensitizer, i.e. it induces apoptosis and necrosis in cells upon irradiation with red light through formation of singlet oxygen. We demonstrated that PDT in vitro and in vivo using cyano-aryl porphyrasine macrocycles is accompanied by a significant viscosity increase by monitoring the fluorescence lifetime of the rotor. We suggest that this increase could be used as a completely new type of diagnostic and dosimetry tool during a PDT treatment providing feedback information about individual therapy status. In addition, the results of in vivo experiments showed that PDT sensitizers prepared on the cyano-aryl porphyrazine pigment platform bound to gadolinium cation demonstrate the potential to become an extraordinarily effective multimodal agent for theranostics, representing a new approach to PDT based on real-time monitoring of the therapy in combination with precise MRI /fluorescence diagnostics of tumours.
Imene SEHOUT
University of Constantine, Algeria
Title: New Green Synthesis of 1.8-Dioxodecahydroacridine Derivatives and Polyhydroquinolines, via Two Natural Catalysts in a Solvent-Free Medium

Biography:
Imene Sehout has her PhD in organic chemistry at the age of 26 years, after years of experience in research, evaluation, teaching in the University of the Brothers Mentouri Constantine- Algeria; her researches were based on the use of new green and safe methods in organic synthesis. She has used natural catalysts to achieve biologically active molecules.
Abstract:
Requirements of modern chemistry, including the development of new synthesis with new secure, economic and environmentally protocols guide us to the field of green chemistry [1]. The development of solvent-free organic synthetic methods has become an important research area, aiming to save energy and to prevent hazardous solvent waste and toxicity in chemical processes. On the other hand, the catalyst is the most important factor in organic synthesis for its important role in a reaction by its presence or by its intervention. Green chemistry has allowed the emergence of new horizons in the field of catalysis; it is now the ninth principle of green chemistry. Several green methods were applied as microwave [2], ultrasound [3], ionic liquids [4], nanoparticles [5]. On our part we have contributed to this effect by the development of two new natural catalysts, inexpensive, non-toxic and very available, which are ascorbic acid and acetyl salicylic acid. We applied our two new natural catalysts in the Hantzsch reaction (Figure:1), which is a multicomponent reaction, it is considered as a green reaction, by the fact they generate in record time and with good yields molecular structures with great complexity. The achieved results are remarkable, we could synthesize a range of 1.8-dioxodecahydroacridine derivatives (6), and polyhydroquinolines (7) with yields ranging between (47-99) % in the time range from (1.5-6) hours, we also enriched the bibliography molecules by synthesis of authentic molecules.
Fabio Luiz Paranhos Costa
Federal University of Goiás, Brazil
Title: Very fast and surprisingly accurate GIAO-mPW1PW91/3-21G//PM7 scaling factor for 13C NMR chemical shifts calculation

Biography:
Abstract:
Fabio Marinelli
University of L’Aquila, Italy
Title: Synthesis of indole derivatives through transition metals-catalyzed cyclization of 2-alkynylaniline derivatives

Biography:
Fabio Marinelli obtained his degree in Chemistry at the University of Rome in 1980. In 1983 he became Organic Chemistry Researcher and, since 2001, he has been Associate Professor of Organic Chemistry at the University of L’Aquila. His research topics include the application of transition metals as catalysts in the synthesis of heterocycles such as indoles, quinolines, lactones and other. He has published more than 115 papers in reputed journals and is member of International Society of Heterocyclic Chemistry.
Abstract:
Indole derivatives are one of the most extensively studied class of heterocyclic compounds. The indole nucleus is a fundamental constituent of many natural and synthetic products with biological activity. Moreover, fused indole derivatives display a number of interesting pharmacological properties. Cyclization of 2- alkynylanilines catalyzed by transition metals represents a powerful tool for the build-up of the indole nucleus, and affords mainly 2-substituded indoles, although sequential functionalizations of nucleophilic 3-position have been reported. An useful development of this methodology is represented by Pd-catalyzed reaction of 2-alkynyltrifluoroacetanilides with a variety of organic electrophiles such as aryl, heteroaryl, alkyl and alkynyl halides, vinyl triflates, arenediazonium tetrafluoroborates, boronic acids, a-iodoenones and allyl esters. This approach is based on the activation of the triple bond towards intramolecular nucleophilic attack by –NHCOCF3 by means of coordination to an organopalladium(II) intermediate; sequential reductive elimination results in the formation of 2,3-disubstituted indoles bearing the organic moieties linked to the -3 position). Furthermore, this methodology allows a straightforward assembly of polycyclic indole derivatives such as indoloquinazolines, indoloquinazolinones, and indoloquinolines.
Ali Karimi
Shahrekord University of Medical Sciences, Iran
Title: In vitro antiproliferative, antioxidant, and apoptosis-inducing activities of dried flower buds of clove extract on human gastric carcinoma

Biography:
Abstract:
Cancer cell resistance to widely used chemotherapeutic agents is gradually developed. Natural products, mainly isolated from medicinal plants, have been considered as valuable sources for herbal anticancer drugs. The present study aimed to evaluate in vitro antiproliferative, antioxidant, and apoptosis-inducing activities of the crude ethyl alcohol extract of dried flower buds of clove (Syzygium aromaticum L.) extract on human gastric carcinoma (AGS). Crude ethyl alcohol extract of dried flower buds of S. aromaticum was prepared. In vitro antiproliferative activity of the extract in AGS and normal (HDFs) cell lines was evaluated using MTT assay. To determine the induction of apoptosis, AGS cells were incubated with one time IC50 concentrations of the extract, stained with both propidium iodide (PI) and Annexin V-fluorescein isothiocyanate (FITC), and analyzed by flow cytometry. Antioxidant activity, total phenolic, and flavonoids content was evaluated with 2, 2-diphenyl-1-picrylhydrazyl (DPPH) assay, Folin-Ciocalteu method and aluminum chloride colorimetric method, respectively. Our results showed that the IC50 of DPPH radical, total phenolic and flavonoid amounts of the extract was 10.05±0.8μg/ml, 225.6±4 mgGAE/g and 29.3±2.35mgRUT/g, respectively. The extract inhibited the proliferation of AGS cells, with IC50 values of 118.7 μg/ml at 48 h after treatment. The results of flow cytometric analysis showed that the extract induced cell apoptosis, with the apoptosis ratio of 21.61% in AGS cell line. In conclusion, the crude ethyl alcohol extract of clove had the best antioxidant activity and the highest total phenolic content and suppresses the proliferation of human gastric cancer cells due to induction of apoptosis.
Alvaro Escribano
Universidad Carlos III de Madrid, Spain
Title: Score-driven dynamic patent count panel data models

Biography:
Abstract:
Tarek Aboul-Fadl
Assiut University, Egypt
Title: Novel Synthetic Inhibitors of Eosinophils with Potential Anti-asthmatic Activity

Biography:
Dr Tarek Aboul-Fadl received his Ph.D. in Pharmaceutical Medicinal Chemistry from Assiut University/Egypt (1994) under the channel system and joint supervision scheme between Assiut University and Josai University/Japan. He performed his postdoctoral training as a postdoctoral research fellow and Scientist at Institute of Pharmaceutical Chemistry, University of Vienna, Austria (1997- 1998), Institute of Pharmacy and Food Chemistry, University of Erlangen-Nürnberg, Germany (1999 and 2013) and Department of Medicinal Chemistry, University of Utah, USA (2001-2002 and 2004-2005). He joined Department of Medicinal Chemistry as an assistant Prof. in 1994, then promoted to associate Prof. in 1999 and to Professor in 2004. He has published more than 75 papers in reputed journals and serving as an editorial board member of repute. He has also registered 4 patents.
Abstract:
Asthma is a major public health issue with high and increasing prevalence rates and a concomitant increase in morbidity and mortality. Asthma is estimated to affect 300 million people, with an expected increase to 400 million worldwide by 2025. Many factors may have contributed to the rise of the problem of bronchial asthma. Increasing air pollution, fast modernization, and widespread construction work are some of the reasons for asthma to thrive. The situation is complicated by poor access to medical services and high price of effective drugs. Asthma is a chronic inflammatory condition, triggered by environmental factors in genetically predisposed individuals, and is characterized by mast cell, T lymphocyte, and eosinophil infiltrates in the bronchial mucosa. Eosinophils are recruited to sites of specific inflammatory reactions, especially during allergic diseases and are correlated with asthma severity. In spite of their numerous adverse effects inhaled glucocortocoids have been established as the standard treatment for asthma. Therefore, an urgent need exists for alternative treatments to overcome these undesirable side effects of steroid therapy and to provide another effective agent for the treatment of asthma. Lidocaine inhibits interleukin-5 (IL-5)-mediated survival and activation of human eosinophils, and it is able to replace inhaled glucocorticoids for the treatment of asthma; however, lidocaine has many undesired side effects mainly due to its sodium channel activity including anesthesia. Accordingly, the current work aims to modify lidocaine structure to obtain analogs with minimum sodium channel IL-5 inhibitory activity. The hypothesis supported by ligand-based pharmacophore modeling generated using different molecular modeling programs.
Ivan Tikhonov
P.G. Demidov Yaroslavl State university, Yaroslavl, Russia
Title: Nitroxyl radicals as antioxidants against lipid peroxidation: an in vitro study

Biography:
Ivan Tikhonov has completed his PhD at the age of 25 years from P.G. Demidov Yaroslavl State University. He is the senior lecturer in P.G. Demidov Yaroslavl State University. He has published more than 10 papers in reputed journals.
Abstract:
Nitroxyl radicals (>NO·) exhibit a set of unique properties (ease of single-electron transfer, paramagnetic properties, cell permeability, antioxidant activity), which makes them attractive for use as drug components. It is known that >NO· catalytically dismute the superoxide anion, inhibit the oxidation of lipids, protect DNA from radicals. We investigated the mechanism of antioxidant action of nitroxyl radicals using a simplified model of lipid membrane (methyl linoleate (LH) in micelles). It has been established that >NO· effectively inhibit azo-initiated LH oxidation even at concentrations of ~10–6 M. Antioxidant activity of >NO· increases with increasing its lipophilicity. Each >NO· radical breaks 2-5 oxidation chains, i.e. the regeneration of antioxidant takes place. This effect is explained by the reaction >NO· + HO2· ® >NOH + O2, while HO2· radical is formed upon the LH oxidation in micelles. This conclusion is confirmed by the results of superoxide dismutase enzyme effect on the kinetics of the process: the antioxidant activity of >NO· decreases, and one >NO· radical terminates one oxidation chain, since the >NO· and HO2· interaction is eliminated. The piperidine-based radicals with a low reduction potential of the oxoammonium cation / nitroxyl radical pair, in particular 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), have the greatest antioxidant activity during the oxidation of LH in the presence of a source of O2·–/HO2· radicals. This results can be useful for the selection of >NO· structures for testing as drug components to protect against the oxidative stress.
The research is supported by RSF grant No. 14-23-00018.
Margitta Dathe
Leibniz Forschungsinstitute of Molecular Pharmacology, Germany
Title: The secrets and potential of a novel cyclic antimicrobial and cell penetrating peptide

Biography:
Margitta Dathe studied physics at the Humboldt University of Berlin and completed her PhD in 1978 from the Academy of Sciences of the GDR. Since 1999 she has been working as head of the peptide-lipid interaction research group of the Leibniz Researchinstitute of Molecular Pharmacology. Her research interest is focused on targeting, cellular uptake promoting and antimicrobial peptides. She has published more than 100 papers in reputed journals.
Abstract:
The development of antimicrobial peptides as antibiotic agents requires structural characterization and understanding of their diverse mechanisms of action. We investigated small cyclic anginine (R)- and tryptophan (W)-rich peptides characterised by variations in the amino acid position, exchange of R and W by other charged or aromatic residues, introduction of D-amino acid residues and reduction and enlargement of the ring size. The cyclic hexapeptide cycloRRRWFW (cWFW) revealed high antimicrobial activity and proved to be not toxic against eukaryotic cells. Its amphipathic structure and arginine content provide the prerequisites for membrane permeabilisation and translocation as modes of action [1-3].Using a number of techniques to study peptide interaction with bacterial and eukaryotic cells and model membranes, we could show that the activity of cWFW is based on a novel antimicrobial mechanism. Strong interactions with the bacterial membrane lead to reduction in membrane fluidity and disturbance of the native lipid matrix. The formation of distinct lipid domains is related to a severe disturbance in the positioning of functional proteins [4]. Chemical modifications such as enhancement of the peptide hydrophobicity or enlargement of the cycle eliminated the bacterial selectivity and induced a membrane permeabilising mode of action. Although cWFW does not enter the cytoplasm of bacteria, it is rapidly internalized into human cells. The combination of cell penetrating properties with high antimicrobial activity and the novel mechanism of action render the cyclic hexapeptide an eligible compound with regard to the treatment of intracellular bacterial infections.
Catherine GUILLOU
Unversité de Paris-Saclay, France
Title: New potent MKLP2 inhibitors in the paprotrain series with high anticancer activity in vivo : Design, Synthesis and Structure-Activity relationships

Biography:
Research activities developed within Catherine Guillou’s group are centred on the development of new methodologies, their application to the total synthesis of biologically active natural products and the design of enzyme inhibitors involved in cancer and Alzheimer’s disease. She is co-authors of 58 publications and 8 patents.
Abstract:
Kinesins constitute a superfamily of microtubule-based motor proteins with important cellular functions ranging from intracellular transport to cell division. Some kinesin family members function during the mitotic phase of the eukaryotic cell cycle and are crucial for the successful
progression of cell division. MKLP-2 (also known as Kif20A, RabK6, RB6K, Rab6KIFL, Rabkinesin6) a member of the kinesin-6 family plays an essential role during cytokinesis and is overexpressed in various
cancers such as pancreatic cancer, bladder cancer, breast cancer, small-cell lung cancer, hepatocarcinogenesis, melanoma and gastric cancer. Furthermore, MKLP-2 is weakly detectable or absent in the normal spleen, lymph nodes, pancreas, lung, brain, liver, kidney and skeletal muscle. MKLP2 is involved in the relocation of chromosome passenger complex CPC (which consists of Aurora B, INCENP, surviving and borealin) to the spindle midzone. Down regulation of MKLP2 inhibits the growth of gastric and pancreatic cancer cells. We recently identified one compound (named paprotrain) as the first known inhibitor of MKLP-2. Paprotrain does not inhibit others members of the kinesin superfamily involved in mitosis. The synthesis, the structure-activity relationships and the biological activities will be discussed.
Lilia Clima
Centre of Advanced Research in Bionanoconjugates and Biopolymers, Romania
Title: Dynamic combinatorial approch as synthetic strategy for the formation of non-viral vectors for gene therapy.

Biography:
Abstract:
The next level in Drug Discovery is the easy building and self-generation of multifunctional nanostructures from commercially available or “easy to prepare” units, which will further self-assemble in a complex, tunable and multifunctional materials, suitable for very specific targeted drug delivery. Many functional platforms have been rationally designed with the hope of mimicking the complicated DNA histone machinery. However, DNA and target cells are highly variable and rational design is limited to a relatively small number of components and a high number of synthetic steps. One possible solution to this problem is to employ a dynamic screening approaches. In presented work adaptive dynamic vectors based on polyethylene glycol, cationic moiety components and in some cases squalene derivative, which are reversibly connected to core centres are prepared and tested as vectors for DNA transfection. Depending on their tuneable composition, these modular vectors dynamically self-adapt to their DNA targets, allowing the rapid screening of most effective vectors, optimally matched to DNA 3D surrounding space. Our strategy allows easy and efficient identification of adaptive vectors with high DNA complexation ability, good transfection efficiency, and well tolerated by mammalian cells.
This work was supported by Horizon 2020 WIDESPREAD 2-2014: ERA Chairs Project no 667387 and a grant of the Romanian National Authority for Scientific Research and Innovation, CNCS/CCCDI – UEFISCDI, project number PN-III-P3-3.6-H2020- 2016-0011, within PNCDI III.
Mustapha A. Tijjani
University of Maiduguri, Nigeria
Title: Isolation and Structural Elucidation of 20 hydroxyecdystone from Vitex doniana Sweet Stem bark

Biography:
Abstract:
The air dried sample V. doniana after collection and identification, was extracted with ethanol and further partition with chloroform, ethyl acetate and n-butanol. The ethanolic extract (11.9g) was fractionated on a silica gel accelerated column chromatography using solvents such as n-hexane, ethyl acetate and methanol. Each eluents fractions (150ml aliquots) were collected and monitored with thin layer chromatography. Fractions with similar Rf values from same solvents system were poled together. Phytochemical test of all the fractions were performed using standard procedure. Complete elution yielded 48 fractions (150ml/fraction) which were pooled to 24 fractions base on the Rf values. It was further recombined and 12 fractions were obtained on the basis on Rf values and coded Vd1 to Vd12 fractions. Vd8 was further eluted with ethylacetate and methanol and gave fourteen 14 sub fractions Vd8-a, -Vd8-m .Fraction Vd8-a (56mg) has gave a white crystal compound coded V1. It was further checked on TLC and observed under Ultraviolet lamp and was found to give a single spot. The Rf values was calculated to be 0.433. The melting point was determined using Gallenkamp capillary melting point apparatus and found to be 241-243°C uncorrected. Characterization of the isolated compound coded V1 was done using FT-infra-red spectroscopy, HNMR, 13CNMR(1and 2D) and HRESI-MS. The IR spectrum of compound V1 shows prominent peaks that corresponds to OHstr (3365cm-1) and C=0 (1652cm-1) etc. This spectrum suggests that among the functional moiety in compound V1 are the carbonyl and hydroxyl group. The 1H NMR (400 MHZ) spectrum of compound V1 in DMSO-d6 displayed five singlet signals at δ 0.72 (3H, s, H-18), 0.79 (3H, s, H-19), 1.03 (3H, s, H-21), 1.04 (3H, s, H-26), 1.06 (3H, s, H-27) each integrating for three protons indicating the five methyl functional groups are present in the compound. It further showed a broad singlet at δ 5.58 integrated for 1 H is due to an olefinic H-atom adjacent to the carbonyl carbon atom. Three signals at δ 3.10` (d, J = 9.0 Hz, H-22), 3.59 (m, 1H, 2H-a) and 3.72 (m, 1H, 3H-e) each integrating for one proton is due to an oxymethine protons indicating that three oxymethine H-atoms are present in the compound. These all signals are characteristic to the ecdysteroid skeletons. The 13C-NMR spectrum showed the presence of 27 Carbon atoms, suggesting that may be steroid skeleton. The DEPT-135 experiment showed the presence of five CH3, eight CH2, and seven CH groups, and seven quaternary C-atoms. The Molecular formula was established as C27H44O7 by High resolution Electron spray ionization-Mass spectroscopy (HRESI-MS) positive ion mode m/z 481.3179. The signals in Mass spectrum 463, 445, and 427 peaks corresponding to losses of one, two, three, or four water molecules are characteristic for Ecdysterone skeleton reported in the literature. Based on the spectral analysis (HNMR, 13CNMR, DEPT, HMQC, IR, HRESI-MS) the compound V1 is thus concluded to have ecdysteriod skeleton and conclusively conforms with 2β, 3β 14α, 20R, 22R, 25- hexahydroxy-5 β cholest-7-ene-6- one, or 2, 3,14, 20, 22, 25 hexahydroxy cholest-7-ene-6-one commonly known as 20-hydroxyecdysone.
Haroon ur Rashid
Sarhad University of Science & Information Technlogy, Pakistan
Title: Mesoporous Nanoparticles as Multimodal Imaging Agents for Magnetic Resonance, X-ray Tomography and Photoluminescence Imaging

Biography:
Haroon ur Rashid has completed his PhD at the age of 32 years from School of Chemistry & Chemical Engineering, Huazhong University of Science & Technology, Wuhan, China and postdoctoral studies from Institute of Chemistry, Federal Univewrsity of Mato Grosso do Sul, Campo Grande, MS, Brazil. He is currently working as Assistant Professor in Department of Chemistry, Sarhad University of Science & Information Technlogy, Peshawar, Khyber Pakhtunkhwa, Pakistan.. He has published more than 14 papers in reputed journals.
Abstract:
Lanthanide-doped Gadolinium nanoparticles have attracted considerable attention due to their promising applications in biological imaging. Sodium gadolinium fluoride (β-NaGdF4) acts as a perfect host material for doping of luminescent lanthanide ions. Presence of seven unpaired electrons in Gd+3 ion, its large magnetic moment and long electronic relaxation time makes it an ideal candidate to enhance water proton relaxation rates. Therefore, Gd-based nanoparticles may also be applied as MRI contrast agents. Due to heavy nature of Gadolinium, it can strongly absorb X-ray radiations. In this work, sodium gadolinium fluoride is doped with ytterbium and erbium to produce β-NaGdF4:Yb/Er as core nanoparticles. They are subsequently coated with sodium gadolinium fluoride doped with neodymium (β-NaGdF4:Nd) to produce β-NaGdF4:Yb/Er@β-NaGdF4:Nd core-shell nanoparticles. Dynamic Light Scattering (DLS), X-ray Diffraction (XRD) and Transmission electron microscopy (TEM) techniques were used for the characterization of nanoparticles. Gadolinium-based nanoparticles doped with Yb and Er or Yb and Tm have proved to be useful up-converting materials. Nanoparticles reported in this work are expected to be useful trimodal contrast agents for Photoluminescence, X-ray tomography (CT), Magnetic Resonance Imaging (MRI).
Mrinalkanti Kundu
TCG Lifesciences Pvt. Ltd., India
Title: Modeling, Synthesis and Biological Evaluation: Identification of Novel Small Molecule Drug-like Anti-cancer Therapeutics

Biography:

Abstract:
Volodymyr V. Tkach
Chernivtsi National University, Ukraine
Title: Theoretical Investigation of the use of CoSn(OH)6 cubic nanoparticles for uric acid electrochemical detection in vivo and in vitro

Biography:
Volodymyr Tkach, 28 years old, has completed his PhD at the age of 25 years from Chernivtsi National University, Ukraine and postdoctoral studies from Federal University of Mato Grosso do Sul, Brazil. He speaks Ukrainian, Russian, English, Portuguese and Spanish. He has published more than 80 papers in reputed journals and his scientific interests includes the development of new conducting polymers and composites for pharmaceutical compounds’and pesticides’ analysis, like also the electroanalytical investigation of cobalt-based materials and their composites with conducting polymers and carbon materials.
Abstract:
Uric acid is the final product of the genetically determinated process of purine metabolism in the human and vertebrate animal organism. Its lack leads to the hyperuricemia and pathological states like Wilson and Fanconi disease. On the other hand, its excess leads to the Lesch-Nyhan syndrom. It makes actual the task of the seek of the efficient mechod of its concentration determination in organism. The redox activity of the uric acid let us apply the electroanalytical methods to it, especially those using the chemically modified electrodes, as the use of the modifiers accelerates the process and makes it more efficient. The cobalt(II)-tin(IV)-hydroxide is an interesting and modern electrode modifier, recently obtained in cubic nanopearticles. It is seen as an interesting candidate to catalyse the photo, photoelectro and electrochemical systems (including the sensing systems). Its use for the detection of the substances, which are oxidized in the similar conditions let us investigate theoretically the determination the possibility of the electrochemical detection of uric acid over an electrode, modified with this material in this work. The behavior of this system may be described by a tridimensial equation set, whose analysis shows that the electroanalytical system is driven efficiently, and may be diffusion- or reaction-controlled. The material may be used solely or in a composite with conducting polymers (polyaniline, polypyrrole, polycarbazole etc) and carbon materials (especially carbon paste, MWCNT and graphene). The oscillatory behavior is possible and may only be caused by DEL-capacitances change.
Markus R. Heinrich
Friedrich-Alexander-Universität Erlangen-Nürnberg, Germany
Title: New synthetic methods for medicinal chemistry

Biography:
M. Heinrich has completed his PhD at Ludwig-Maxilians-Universität München (Germany) in 2003. After postdoctoral research at Ecole Polytechnique (Palaiseau/France) and habilitation at Technische Universität München (2009) he was appointed as a professor for pharmaceutical chemistry at the Friedrich-Alexander-Universität Erlangen-Nürnberg (Germany). He has published more than 75 papers in reputed journals and he currently serves as a speaker of the Emil Fischer Graduate school at the FAU.
Abstract:
Our research group is interested in developing new radical reactions with a strong focus on a later application in the field of medicinal chemistry. Key reaction types are alkene and arene functionalizations. While important contributions could be made in the area of the Meerwein arylations (carboamination, carbooxygenation, carbofluorination), our efforts in radical aryl-aryl coupling are mainly dedictated towards the improvement of reactivity and selectivity as well as the late stage functionalization of peptides. The recent discovery of phenylazocarboxylates as versatile bifunctional reagents not only offers attractive synthetic pathways regarding the preparation of 18-fluorine-labeled radiopharmaceuticals, it also provides multiple options for combinatorial synthesis. Moreover, we have been able to show that important building blocks for pharmaceuticals can be prepared in a highly sustainable way using nitrogen oxides. So far, our newly developed reactions have been applied for the synthesis of antimalarials, numerous GPCR ligands, enzyme inhibitors as well as diverse radiolabeled compounds for PET imaging.
Yusuf M. Al-Hiari
The University of Jordan, Jordan
Title: Novel Potential Antihyperlipidaemic Agents Derived From Heterocyclic-2-carboxamides

Biography:
Yusuf Al-Hiari has completed his PhD at the age of 31 years from Strathclyde University/UK and then appointed as assistant prof. At The faculty of Pharmacy, JU. He was promoted to full Prof. During 2012. He has published more than 65 papers in reputed journals with impact factor, 6 patents and has been serving as an editorial board member of repute journals.
Abstract:
More than 5 heterocyclic systems were revealed with potential hypolipidemic activity by our group such as indole, pyrrole, furan, benzofuran and pyridine. All derivatives prepared holds carboxamide attached to highly lipophilic substituents such as benzophenones and antharaquinones. This presentation aims to prepare new 5-substituted indole-1H-indole-2-carboxamide derivatives I as a model and to investigate their hypolipidemic activity in-vivo using Triton WR-1339-induced hyperlipidemic rats as animal model. The novel compounds involve 5-Fluoro-1H-indole-2-carboxamide derivatives of benzophenones1 (series A; 5, 6, 12-15), anthraquinones2 (series B; 17, 19), anilines (series C; 21, 25) and acetophenones (series D; 26-27). The new derivatives represented by compounds 5, 6, 17m and 19 have shown a significant reduction in triglyceride levels (69-90%) compared to hyperlipidemic rats. The analogous 5-Chloro , 5-Bromo and 5-methoxy-1H-indole-2-carboxamide derivatives did not change the pattern of activity with all active derivatives. The elevated plasma triglyceride levels produced by Triton WR-1339 administration were significantly (p < 0.0001) suppressed in bezafibrate (79%), in compound (5) (90%), in compound (6) (83%), in compound (17m) (69%) and (79%) in compound (19) after 12 h in comparison to hyperlipidemic control (HG). At the same time, high-density-lipoprotein cholesterol levels were significantly increased after 12 h of Triton administration (+ 86% p < 0.0001) in compound (5), (+ 53% and +27% p<0.01) in compound (6), BF and (+ 16% and + 37% p<0.05) in compound (17m) and (19) respectively compared to hyperlipidemic control (HG).
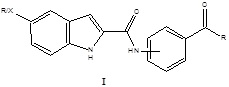
Christian Cavé
University Paris Sud, France
Title: Design and synthesis of inhibitors of the GDP-Mannose Pyrophosphorylase(GDP-MP)

Biography:
Christian Cavé has completed his PharmD at the age of 22 years from University of Paris Sud and this PhD at the age of 29 years from University of Paris Sud (France). In 1997 he was full professor of Organic chemistry at the University of Burgandy then since 2002 he is full professor at the Faculty of Pharmacy, Uniserty of Paris Sud. He was director of a research team focusing on the asymmetric synthesis and Medicinal Chemistry and currently he works in the team "Chimiotherapie Antiparasitaire" UMR CNRS BioCIS.
Abstract:
Leishmaniasis is a neglected tropical disease who occurs in 88 countries and infects 12 million people worldwide that causes hundreds of thousands of deaths per year. Only few treatments are currently available (i.e. antimonials, miltefosine and AmBisome®) while more and more cases of drug resistance appear all around the world. To overcome the limitations of these treatments, we have started to develop a new series of inhibitors of an original target that are essential for parasite survival or virulence : the GDP-Mannose Pyrophosphorylase(GDP-MP), an enzyme involved in glycosylation and essential for amastigote survival. The goal of this project is to develop inhibitors that are specifically active on the GDP-MP of Leishmania infantum and do not have side effects on the human homologous enzyme. Currently we are working on click chemistry in order to synthesize stable analogs of GDP-MP in Medicinal Chemistry,
Roman Pliss
P.G. Demidov Yaroslavl State University, Russian Federation
Title: Influence of solvation on the mechanism of the hydroperoxide radicals' addition to the π-bonds of 1,2-and 1,4-diphenylbutadiyne-1,3

Biography:
Roman Pliss has finished Yaroslavl State Technical University (Yaroslavl, Russia) in 1999. He has published more than 10 papers in reputed journals.
Abstract:
The role of the effect of nonspecific solvation in the addition of hydroperoxide radicals to π-bonds of trans-1,2-diphenylethylene and trans,trans-1,4-diphenylbutadiene-1,3 in the medium of different polarity solvents was investigated via a set of kinetic methods (microvolumetry, method of inhibitors, rotating-sector method). When the medium polarity is varied, the logarithm of the rate constant of this reaction in the Kirkwood-Onsager equation coordinates shows a linear dependence. Quantum-chemical analysis (NWChem, DFT B3LYP/6-311G**) allows us to conclude that the influence of the polarity of the medium for nonspecific solvation on the rate of the considered reaction will depend on how much the solvation energies of the transition complex and the initial reaction complex differ. If the solvation energy of the transition complex is higher than the solvation energy of the initial reaction complex, the reaction rate should increase with increasing solvent polarity, otherwise it should decrease.
The research is supported by RSF grant No. 14-23-00018.
Satinder Ahuja
Ahuja Consulting, USA
Title: New drug development and problem-solving in the pharmaceutical industry

Biography:
Dr. Satinder Ahuja successfully managed development of many new drugs at Novartis Corporation for over 25 years in a number of leadership positions that led to earnings of billions of dollars. His expertise is recognized worldwide in chromatography and separations, especially in the area of ultratrace analyses, chiral separations, and characterization of contaminants by various techniques. As President of Ahuja Consulting, he has advised major pharmaceutical companies in the US and abroad on new drug development and on green chemistry solutions to environmental issues. He has given plenary lectures worldwide, won awards, and published numerous papers and over twenty books.
Abstract:
New drug development may take as many as nine years and requires input from medicinal chemistry, analytical chemistry, chemical development, pharmaceutics, pharmacology, toxicology, manufacturing, marketing, and regulatory departments. The inputs from Analytical R&D are important for all of these operations because they can make the difference between slow and fast development. Drug discovery is generally initiated with the synthesis of a new chemical entity (NCE) based on combinatorial chemistry, or drugs are based on recombinant products. The molecular structure, including chirality, has to be confirmed. It is necessary to demonstrate the absence of any undesirable impurities, including enantiomers that may exhibit unusual pharmacologic or toxicologic activities (1–6). Finally, it is necessary to select an optimum dosage form, based on therapeutic and marketing needs. The selected dosage form has to meet GMP/GLP requirements. Discussion will focus on how various advances in chromatography and spectroscopy can help new drugs development and provide green chemistry-based solutions to a variety of problems encountered in the pharmaceutical industry.
Carlos Henrique Tomich de Paula da Silva
University of São Paulo, Brazil
Title: Discovery of novel aryl hydrocarbon receptor (AhR) antagonists using homology modelling and ligand-based drug design

Biography:
Carlos Tomich has completed his PhD at the age of 31 years from University of São Paulo and postdoctoral studies from University of São Paulo and PRBB, at Barcelona-Spain. He is the Associate Professor of Pharmaceutical Chemistry at University of São Paulo, with a drug design research position. He has published more than 120 papers in reputed journals and has been serving as an editorial board member of repute.
Abstract:
Aryl hydrocarbon receptor (AhR) is a transcription factor activated by ligand. However, AhR also modulates many physiological and pathological processes that affect inflammatory and immunological responses. There is a growing interest in discovery of selective competitive antagonists for AhR, where the most potent ones exhibit acceptable antagonist properties but they also show partial agonist activity or exhibit agonist activity on estrogen receptors (ER). Also, limited availability of selective and pure competitive AhR antagonists and scarce structural information regarding AhR binding domain (located in the PAS-B domain) are reported. In this work, a preliminary search in the Protein Data Bank (PDB) for AhR structures revealed only one – a PAS-A domain, but not PAS-B. A homology model of the PAS-B domain of the AhR receptor was then carried out, using PAS-B structures of ARNT as templates. A flexible docking approach was then used with the most potent AhR antagonists reported, allowing us to derive (and to validate) a pharmacophoric pattern common to the compounds thus aligned. Two subsequent virtual screening experiments were then performed in databases of commercially available compounds, using: (1) the pharmacophore model; (2) the shape and electrostatic potential of the most potent AhR antagonist reported. In sequence, 29 compounds were filtered regarding to both atoxicity and good pharmacotherapeutic profile, thus predicted in silico. Finally, at least 5 novel atoxic AhR antagonists have been discovered, which experimentally showed atheroprotective efficacy correlated to the AhR antagonism, since they inhibited, almost completely, AhR-mediated oxLDL uptake by murine macrophages, induced by TCDD.
Brandi M. Baughman
National Institute of Environmental Health Sciences, USA
Title: A High-Throughput Screening-Compatible Strategy for the Identification of Inositol Pyrophosphate Kinase Inhibitors

Biography:
Abstract:
Sandrine ONGERI
Université Paris Saclay, France
Title: Designed peptidomimetics disrupt protein-protein interactions mediating amyloid protein aggregation

Biography:
Sandrine Ongeri has completed her PhD in 1999 from Université Paris Descartes (France) and postdoctoral studies from the University of Milano (Italy). During her PhD, she spent one year at Oxford University (UK). She is group currently leader at the University Paris Sud. Her group focuses on the design, the synthesis and biophysical evaluations of peptidomimetics as inhibitors of protein-protein interactions involving beta-sheet structures, in particular in amyloid proteins aggregation. Her group's research is also expert in the synthesis of unantural fluorinated amino acids and in the use of fluorine as probe in peptide-protein interaction.
Abstract:
Amyloid fibrils are self-assembled insoluble aggregates that constitute the hallmark of more than 20 serious human amyloidosis diseases, such as Alzheimer’s disease (AD) and type II diabetes. Current drugs have failed to slow the progression of AD, which affects more than 35 million people worldwide. How drug candidates that reduce fibril formation act on the most neurotoxic oligomeric forms of amyloid peptide Aβ1-42 is far from being established. We report herein the capacity of two new classes of peptidomimetics to inhibit both Aβ1-42 early oligomerization and fibrillization: 1- sugar-based peptidomimetic analogs having physicochemical properties for drug-likeness[1]; 2- β-hairpin mimics[2]. A wide range of bio- and physico-chemical techniques, such as Thioflavin-T fluorescence spectroscopy, transmission electronic microscopy, a new developped capillary electrophoresis method[3], electrospray differential mobility analysis[4], nuclear magnetic resonance, and surface plasmon resonance, was used in order to identify the molecular mechanisms by which these new series of molecules can delay the aggregation of Aβ1-42. This is the first example of small molecules that preserves the non toxic monomeric species of Aβ1-42. Some compounds suppress totally the toxicity of Aβ1-42 towards SH-SY5Y human neuroblastoma cells, even at sub-stoichiometric concentrations. This protective effect is much more significant than that observed with molecules that have undergone clinical trials which reduce Aβ1-42 toxicity only at stoichiometric or higher concentrations. Preliminary results on the inhibition of IAPP aggregation involved in type II diabetes will be also presented.
Roman Pliss
P.G. Demidov Yaroslavl State University, Russian Federation
Title: Influence of solvation on the mechanism of the hydroperoxide radicals' addition to the π-bonds of 1,2-and 1,4-diphenylbutadiyne-1,3

Biography:
Roman Pliss has finished Yaroslavl State Technical University (Yaroslavl, Russia) in 1999. He has published more than 10 papers in reputed journals.
Abstract:
The role of the effect of nonspecific solvation in the addition of hydroperoxide radicals to π-bonds of trans-1,2-diphenylethylene and trans,trans-1,4-diphenylbutadiene-1,3 in the medium of different polarity solvents was investigated via a set of kinetic methods (microvolumetry, method of inhibitors, rotating-sector method). When the medium polarity is varied, the logarithm of the rate constant of this reaction in the Kirkwood-Onsager equation coordinates shows a linear dependence. Quantum-chemical analysis (NWChem, DFT B3LYP/6-311G**) allows us to conclude that the influence of the polarity of the medium for nonspecific solvation on the rate of the considered reaction will depend on how much the solvation energies of the transition complex and the initial reaction complex differ. If the solvation energy of the transition complex is higher than the solvation energy of the initial reaction complex, the reaction rate should increase with increasing solvent polarity, otherwise it should decrease.
The research is supported by RSF grant No. 14-23-00018.

Biography:
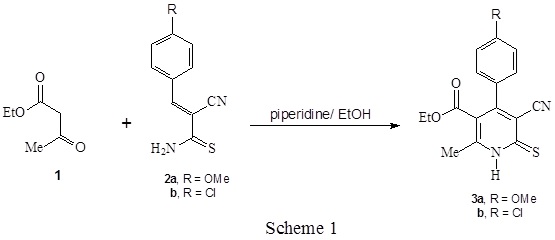
Abstract:
Reaction of 4-aryl-3-cyano-5-ethoxycarbonyl-6-methylpyridine-2(1H)-thiones 3a,b with chloroacetonitrile gave the corresponding 3-aminothieno[2,3-b]pyridine-2-carbonitriles 5a,b. Condensation of 5a,b with triethyl orthoformate produced the methanimidate derivatives 7a,b. Treatment of 7a,b with hydrazine hydrate at room temperature resulted in the formation of ethyl 3-amino-9-aryl-3,4-dihydro-4-imino-7-methylpyrido[3’,2’:4,5]thieno[3,2-d]pyrimidine-8-carboxylates 8a,b. 3-Amino-4-(4-methoxyphenyl)-5-ethoxycarbonyl-6-methylthieno[2,3-b]pyri-dine-2-carboxamide (6) was prepared and reacted with triethyl orthoformate to give pyrimidine-4(3H)-one derivative 15. Chlorination of 15 with phosphorus oxychloride gave 4-chloropyrimidine 16, which in turn was reacted with hydrazine hydrate to produce ethyl 4-hydrazino-9-(4-methoxyphenyl)-7-methylpyrido[3’,2’:4,5]thieno [3,2-d]pyrimidine-8-carboxylate (18). Compounds 8a,b and 18 were used as precursors for synthesizing other new pyridothienopyrimidines as well as triazolopyridothieno-pyrimidines, and pyridothienopyrimidotriazinoindoles. Structural formulas of all newly synthesized compounds were confirmed by elemental and spectral (IR, N MR and mass) analyses.

Biography:
Abstract:
Fana Abdulrahman
University of Maiduguri, Nigeria.
Title: Phytochemistry, Isolations and Some Pharmacological Studies of ethanol leaf extract of Piliostigma thonningii

Biography:
Abstract:
This study was aimed at evaluation of phytochemical constituents and the effect of ethanol leaf extract of Piliostigma thonningii on the central and peripheral nervous systems in laboratory animals. Fresh leaves of Piliostigma thonningii were air-dried, pulverized extracted using soxhlet extraction technique with ethanol 148.24% w/w after being concentrated. The extract was screened for phytochemicals using standard methods. 20 g of the ethanol extract was subjected to column chromatographic (CC) analysis using ethyl acetate and n-butanol as mobile phase at different ratios and silica gel of 60-120 mesh as the stationary phase. Fractions obtained with similar retention factor (Rf) using thin layer chromatography (TLC) were combined, coded and subsequently screened for phytochemicals. Subsequent purification of fraction PTE3 was carried out using CC (ethylacetate and methanol were used as mobile phase at different ratios) and TLC until a sub-fraction PTE34 amongst other fractions gave a single spot on TLC and had a melting point of 102-103 oC. The phytochemical studies of the ethanol leaf extract of Piliostigma thonningii revealed the presence of some useful chemical compounds such as flavonoids, cardiac glycosides, tannins, saponins, and terpenoids. The pharmacological effects of Piliostigma thonningii was determined by examining the effects of the leaf extract on phenobarbitone sleeping time, analgesic and muscle relaxant activities using experimental animals. The analgesic effect of the leaf extract was evaluated with acetic acid induced writhing and thermally induced Nociception for pain. It was observed that the extract conferred 48.00 and 57.20% protection from writhes induced by acetic acid on mice when extract doses of 200 and 400 mg/Kg were administered. Similarly, there was a significant (p<0.5) dose dependent effect conferred on mice when pain was induced by heat. The extract also had a muscle relaxant effect as 20%, 60% and 80% were observed to slide down an inclined board in a dose dependent manner. The extract also significantly potentiated sleeping time of phenobarbitone dose dependently in rats of which the mean time duration of (72.0±04.64) min, (83.40±02.11) min, and (123.60±11.57) min were observed when rats were administered extract doses of 200, 400 and 600 mg/Kg b wt. Thus the ethanol leaf extract of Piliostigma thonningii was able to provide depressant effects which were shown in its ability to potentiate barbiturate sleeping, analgesia and muscle relaxant effect.
Nyane NA
University of KwaZulu-Natal, South Africa
Title: Characterising Chalo-Naringenin Analogs as Putative Therapeutic Agents for Type 2 Diabetes

Biography:
My name is Ntsoaki Anna ‘Nyane ,I am African female aged 25 years old, who studied BPharm Degree at University Of Lesotho. I enrolled in my studies at University of KwaZulu-Natal where I am currently doing my Research under the department of Pharmacology, School of Health Sciences. I am in my second year of Masters. I was awarded first prize (30000-travel voucher) in research symposium 2016 for winner in the Masters oral category held by College of Health sciences. I have also assisted the Honours students with their Laboratory Experiments. I am doing lecturing on third level and fourth level at University of KwaZulu-Natal in school of Pharmacy. I have two accepted manuscript in Journal of Pharmacology and the other is PLOS ONE.
Abstract:
Tove Tuntland
Novartis Institute of Biomedical Research (NIBR), USA
Title: A Modern In Vivo PK Paradigm: Combining Snapshot, Rapid and Full PK Approaches to Support Early Drug Discovery

Biography:
Tove Tuntland holds a Pharmacy degree from the University in Oslo, Norway, and a Ph.D. in Pharmaceutics from the University of Washington, Seattle, USA. She has expertise in preclinical drug metabolism and pharmacokinetics (DMPK), pharmacology and PK/PD, and worked in discovery and development at Pfizer Global Research and Development (PGRD) in La Jolla, California (1996 to 2002). Thereafter until present time she has led a group at Genomics Institute of Novartis Research Foundation (GNF), supporting in vitro and in vivo preclinical DMPK and PK/PD studies in a variety of discovery and development programs in oncology, immunology, infectious and metabolic diseases.
Abstract:
Successful drug discovery relies on selection of drug candidates with good in vitro ADME and in vivo pharmacokinetic properties as well as appropriate preclinical efficacy and safety profiles. However, in vivo animal pharmacokinetic studies are often conducted in a traditional low throughput manner, and therefore, are the bottlenecks of discovery projects in many pharmaceutical companies.
This presentation will focus on the tiered in vivo PK approaches, including snapshot PK, rapid PK and full PK study designs we have implemented to support our drug discovery efforts. In all 3 approaches, compound is dosed and analyzed discretely, thereby eliminating any drug-drug interaction concerns and analysis complications typically associated with cassette dosing or cassette analysis. The rapid PK approach uses several integrated and automated processes and sample pooling strategy to improve throughput, and has become our main stream in vivo PK approach in the lead optimization stage. These in vivo PK approaches differ in throughputs, capacities and the resources required, and are designed to address the varying needs of drug discovery projects at different stages of project progression. These approaches are well integrated within discovery research, allow tremendous flexibility and are highly efficient in supporting the diverse needs and increasing demand for in vivo profiling. Examples of each of the tiered in vivo PK studies will be illustrated.
Romano V.A. Orru
VU University, The Netherlands
Title: Multicomponent reactions: advanced tools for sustainable synthesis of pharmaceuticals

Biography:
Abstract:
Multicomponent reactions (MCRs) receive increasing attention because they address both diversity and complexity in organic synthesis. With these one-pot reactions diverse sets of relatively complex structures, especially heterocycles, can be generated from simple starting materials. In many MCRs (e.g. the Ugi reaction), isocyanides are important building blocks. Recently, isocyanides have found also application as versatile C1 building block in palladium catalysis. These reactions offer a vast potential for the synthesis of nitrogen containing fine chemicals. In this presentation, the development of novel atom- and step efficient Pd-catalyzed reactions involving isocyanide insertion will be presented. Further, in order to address stereoselectivity issues connected to certain MCRs, biocatalysis offers unique opportunities. Recently, we have developed several methods based on the enzymatic desymmetrization of meso-pyrrolidines using a monoamine oxidase N (MAO-N) from Aspergillus niger optimized by directed evolution and its combination with highly diastereoselective Ugi-type three-component and Ugi-Smiles reactions. In this presentation we highlight several aspects of this chemistry in the context of heterocycle synthesis with applications in green chemistry and pharmaceuticals.
Vasily Sen
P.G. Demidov Yaroslavl State University, Russian Federation
Title: Nitroxide-modified chitosans with enhanced antioxidant potential

Biography:
Vasily D. Sen’ has completed his PhD at the age of 26 years from Institute of Organic Chemistry of Russian Academy of Sciences (RAS). Ðе is a head of Laboratory of stabile radicals in Institute of Problems of Chemical Physics of RAS. He has published more than 60 papers in reputed journals and is an author in several patents.
Abstract:
Chitosan is a biocompatible glucosamine polymer with properties suitable for a great variety of biomedical applications. Water-soluble at neutral pH chitosan-(poly)nitroxides (CPNs), differing in molecular weight of the saccharide backbone (Mw ~1 and ~10 kDa) and the nature of nitroxide radical (pyrroline and piperidine), were obtained and characterized. Fractions of modified glucosamines were in the range 0.15 – 0.28. The dependence of delay time of 2,2-azobis-2-methylpropanimidamide dihydrochloride (AAPH)-induced erythrocyte hemolysis on the concentration of CPNs and data of electron paramagnetic resonance (EPR) spectra indicate their binding to the cell membrane. Because of this binding, CPNs demonstrate identical delay times of hemolysis at concentrations ~100 times lower than structurally similar low molecular nitroxides. The proposed mechanism includes multiple breaking of oxidation chain through nitroxide oxidation to oxoammonium cations and reduction of the latter by common biological reductants. Due to easier oxidation of piperidine nitroxide by radicals RO2• to oxoammonium cations, CPNs with attached 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) more effectively inhibit hemolysis than their counterparts with 2,2,5,5-tetramethylpyrroline-1-oxyl. Work is in progress in designing of amphiphilic CPNs and CPNs with negatively charged groups (heparinomimetics), as well as nanostructures based on them. The effect of new materials on the viability of normal and tumor cells under oxidative stress will be investigated, as well as their use for drug delivery and cardiovascular applications.
The research is supported by RSF grant No. 14-23-00018.
Philip C Bulman Page
University of East Anglia, UK
Title: New Systems for Organocatalytic Asymmetric Epoxidation

Biography:
Phil Page graduated from the Department of Chemistry, Imperial College London in 1975, and obtained his PhD from the same institution in 1978 under the supervision of Prof S V Ley. He spent two years working with Prof L A Paquette at Ohio State University as an SERC/NATO Research Fellow before moving to the University of Liverpool as a lecturer in 1983. He moved to the chair of organic chemistry at Loughborough University in 1996, and to the chair of organic chemistry at the University of East Anglia in 2007. He has published over 200 articles, and has won a Nuffield Foundation Science Research Fellowship, the Royal Society of Chemistry Hickinbottom Fellowship, the Royal Society of Chemistry Tilden Medal, the Glaxo-Wellcome Award for Innovative Chemistry, and a Royal Society Industry Fellowship. His research interests lie principally in asymmetric synthesis/catalysis, synthetic methodology and natural product synthesis. Prof Page, a Kentish Man, is married and lives with one wife and three cats. His hobbies include chemistry, reading, hi fi and music, not necessarily all at once, and food and drink.
Abstract:
The development of methods for the introduction of asymmetry into organic molecules remains a topic of great importance. Catalytic systems are particularly desirable, and the combination of a catalytic asymmetric process with an environmentally friendly reaction system and an inexpensive oxidant offers an especially attractive goal. Non-racemic chiral epoxides are important intermediates for enantioselective carbon-carbon bond formation. We are developing organocatalytic systems in which asymmetric oxidants are formed by reaction of iminium salts with simple oxidants under mild conditions. We currently formulate the reactive intermediates as oxaziridinium ions, from which the iminium salt mediators are regenerated following oxygen transfer to alkene substrates. We can accomplish epoxidation of simple alkenes with up to ca 99% ee. Catalyst loading may be as low as 0.1 mol%. The epoxidation reactions may be carried out under aqueous or non-aqueous conditions. The iminium salt mediators can be easily prepared without chromatography in many cases, and the procedures used are simple to carry out, and require no preparation of unstable reagents. The lecture will discuss recent developments including new generations of catalyst, the first examples of kinetic resolution, the use of non-aqueous as well as the usual aqueous conditions, and alternative oxidants in place of Oxone, including hydrogen peroxide, bleach, and even electrochemical conditions by oxidant generation at boron-doped diamond electrodes.

Biography:
Abstract:
This study is aimed at evaluating the biochemical effects and antioxidants activity of extracts of Vernoia calvoana Hook. f (V.C) on STZ induced diabetic rats. Thirty-six (36) rats weighing (100-150g), were divided into 6 groups of 6 animals each. Groups 1 and 2 representing normal and diabetic controls (NC and DC) respectively, receiving placebo, while groups 3-6 represented diabetic treated, receiving 500 mg/kg b.w metformin, 400 mg/kg b.w crude, n-hexane and methanol fractions of V.C, respectively. Treament with drug and extracts of V.C showed a decrease in fasting blod glucose (FBG) in all experimental groups and was significant (p<0.05) on the 7th day of the experimental period, compared to diabetic control. Progressive increase in body weight was observed in all experimental groups compared to DC group. A significant (p<0.05) increase in glutathione peroxidase (GPX) and catalase (CAT) activities were recorded in all experimental treated animal compared to DC and NC. Malondialdehyde (MDA) concentration was observed to decrease significant (p<0.05) in all experimental groups compared to DC. Histopathologically, the changes in pancreatic integrity was consistent with that of biochemical findings. It may be concluded that, extracts of V.C possess potent ameliorative activity against STZ-induced diabetes, via a potential free radical mopping activity.
Mohammed Alqarni
De Montfort University, UK
Title: Mathematical Modelling of polychromatic light photokinetics

Biography:
Abstract:

Biography:
Jovana Bogojeski has completed her PhD diploma in 2012 under the supervision of Prof. Živadin D. BugarÄić at the Faculty of Science, University of Kragujevac and postdoctoral studies at the University of Braunschweig, Germany, in the group of Prof. Matthias Tamm. She is employed at Faculty of Science, University of Kragujevac as an assistant professor and her research activity is based on the study of kinetics and mechanism of chemical reactions of transition metal complexes with different biomolecules. She has published more than 20 papers in reputed journals.
Abstract:
A few novel rhodium(III) complex [RhIII(X)Cl3] (X = pyridin-bis(pirazole) ligands) were synthesized containing a pincer type, tridentate nitrogen−donor chelate system. All complexes were fully characterized, single crystal X-ray structure analysis has been done. The reactivity of the synthesized complex toward small biomolecules (L-methionine (L-Met), guanosine-5’-monophosphate (5’-GMP), L-histidine (L-His) and glutathione (GSH) and to a series of duplex DNAs and RNA was investigated. These measurements showed that the synthesized complex has a good affinity toward studied ligands and the obtained order of reactivity is: 5'-GMP > GSH >
L-Met > L-His. Duplex RNA reacts faster than duplex DNA, while shorter duplex DNA
(15mer GG) reacts faster compared with 22mer GG duplex DNA. In addition, a higher reactivity is achieved with a DNA duplex with centrally located GG-sequence than with 22GTG duplex, in which GG‑sequence is separated by a T base. Furthermore, the interaction of this metal complex to calf thymus DNA (CT-DNA) and bovine serum albumin (BSA) was further examined by absorption (UV-Vis) and emission spectral studies (EthBr displacement studies). Overall, the studied complex exhibited good DNA and BSA interaction ability. All obtained results in this study indicate that the introduction of pincer-type spectator ligand can be used to improve the reactivity of rhodium(III) complexes. Together, these observations show the reactivity characteristics needed for a potential anti-tumor agent, with the ability to target both DNA and proteins. Every new contribution in this field highly is warranted due to the current lack of clinically used metallo-based alternatives to cisplatin.
Fouzia GHERRAK
University of Blida, Algeria
Title: Development of a liposomal suspension to increase the skin penetration of Diclofenac

Biography:
Abstract:
Several formulation strategies for modifying the skin permeability and for potentiating the activity of an active compound can be envisaged. Diclofenac diethylamine is a nonsteroidal anti-inflammatory drug widely prescribed to treat mild and moderate pain, inflammation and osteoarthritis by inhibiting the enzyme cyclooxygenase. It is already marketed in various forms (gel, emulgel, ophthalmic solution, immediate and prolonged release tablet, suppositories and intramuscular injection). Because of its low solubility in water and in the acidic environment of the stomach, Diclofenac has a low availability and short half-life of about two hours. A topical application for the treatment of rheumatic diseases is therefore suggested to increase bioavailability and decrease toxicity. The objective of this work was to develop a liposomal suspension encapsulating a non-steroidal anti-inflammatory which will constitute the dispersed phase of a gel for dermic use. Diclofenac preparations for dermal application are only marketed in the form gel and emulgel. This liposomal form has been proposed in order to increase the skin penetration or even the availability of Diclofenac thanks to the phospholipidic bilayer of the liposome having the same Structure so that the skin can cross the skin barrier (horny layer). The liposomes were prepared from soy lecithin, cholesterol by the reverse- phase evaporation method. Then analyzed by electron scanning microscope, high-performance liquid chromatography, laser particle size and Zeta-metry to determine their size, encapsulation rate and zeta potential. An optimum with a maximum amount of ethanol in relation to water combined with maximum levels of cholesterol and lecithin was obtained, which promotes the production of small-sized liposomes containing a relatively high surface charge (zeta potential) and having only a narrow polydispersity, thus meeting the stability criteria favoring transposition on an industrial scale.
Yin Zhou
The University of British Columbia, Canada
Title: Sizing α, β and γ Cyclodextrins by Capillary Electrophoresis and Indirect UV Detection

Biography:
Yin Zhou is fourth-year undergraduate Chemical Biology student at the University of British Columbia. She has been an analytical Chemistry research assistant in Chen Research Group since September 2015, where she started her own research project to size cyclodextrins. She is also an Organic Chemistry tutor in the same college, science and Math tutor for high school students. She mentored 14 high school students with their science projects. She has been a social Psychology research assistant, being involved in Gerontology since 2015.
Abstract:
Sizing α, β and γ Cyclodextrins by Capillary Electrophoresis and Indirect UV Detection: Cyclodextrins have ring structures made of glucose connected by 1,4-glycoside linkage and they differ in the number of glucose on the ring. The interior of cyclodextrin is considerably less hydrophilic than its exterior; therefore, they are useful for carrying hydrophobic molecules. Due to their hydrophilic exterior, they are able to penetrate body tissues, which makes them good candidates as drug carriers where they can release biologically active compounds under specific conditions. However, different sizes of cyclodextrins form different complex with the same molecule, so, it is important to know the size of each cyclodextrin. Indirect UV-capillary electrophoresis and Taylor dispersion analysis are used to size α, β and γ cyclodextrins. Because cyclodextrins have low UV absorbance, indirect UV is used in which the background electrolyte has significantly higher UV absorbance than the target molecules, therefore, resulting in negative peaks. Using TDA, diameters of α- and γ-CD are calculated to be 0.70nm and 0.86nm. The small standard deviation indicated the precise and reproducible measurement.
Apurba K. Bhattacharjee
Georgetown University, USA
Title: Discovery of novel reactivators for DFP-inhibited acetylcholinesterase using in silico pharmacophore modeling

Biography:
Dr. Bhattacharjee currently an Adjunct Faculty Professor has over 25 years of research experience in quantum chemistry, pharmacophore modeling, and virtual screening of compound databases. His experiences cover drug discovery programs for antimalarials, antileishmanials, insect repellents and counter agents against OP poisoning. Current interests are in the discovery of antivirals targeting dengue, West Nile and Zika viruses. He has authored and coauthored over 120 publications, three book chapters and 5 patents. Prior to joining the Georgetown University, he served in the Walter Reed Army Institute of Research, Silver Spring, Maryland (U.S.A.) as the Chief Molecular Modeler for drug discovery programs.
Abstract:
Organophosphorus (OP) compounds such as tabun, soman, DFP, sarin, cyclosarin and some pesticides are highly toxic group of compounds. Of the OP-agents, tabun (GA) is the most toxic and developing antidotes for it is a challenging problem. However, tabun being a G –simulator, less toxic DFP is frequently used in experiments for discovery of new reactivators. Using in silico strategy from our previously reported pharmacophore model containing a hydrogen bond acceptor, a hydrogen bond donor, and an aromatic ring (Chem Res & Toxicol, 2010, 23, 26-36), we discovered 17 non-oxime reactivators for DFP-inhibited AChE from a virtual screening of in-house database and two commercial databases, Maybridge and ChemNavigator. All these non-oximes contained a nucleophile group in lieu of the oxime moiety and none was reported before to have potent reactivation efficacy. Efficacy (kr) of all the 17 non-oximes were found to be within 10-fold of 2-PAM in an eel DFP-inhibited AChE in vitro assay. One the non-oximes showed in vivo efficacy comparable to 2-PAM against brain symptoms for DFP-induced neuropathology in guinea pigs. Moreover, two of them showed promising results with no major differences in brain restoration of AChE activity compared to 2-PAM after DFP exposure. Since a serious loss of cholinergic function in the central nervous system contributes significantly to the cognitive symptoms associated with AD and other advanced age related brain diseases, these results on competitive OP-inhibited AChE reactivation and inhibition behavior of non-oximes may be useful for further neurological therapeutics studies.

Biography:
Professor Afzal Mohammed holds a personal Chair in Pharmaceutics and has research expertise in paediatric medicines development which includes pharmaceutical excipient characterization, process and technology innovation to develop orally disintegrating tablets. He has over 15 years of experience in developing novel pharmaceutical processes and formulations and is the lead inventor for dry particle coater. He has over 100 publications including research articles, review papers, book chapters and patent applications.
Abstract:
Dry powder coating is a micro particle engineering process and involves the adsorption of “guest” particles onto the surface of “carrier” particles. This process requires conditions that enable frequent contact between guest and carrier particles, particle adsorption, and ultimately lowering of surface energy of the binary mixture. Although the theoretical paradigm for dry-coating is known, progress and pragmatic translation have been limited owing to the lack of processes and devices capable of producing composite particles while maintaining the innate characteristics of both components (guest and carrier). For instance, mechanofusion works on the principle of high centrifugal forces that generate heat thereby limiting its pharmaceutical application to heat labile materials. Similarly, processing materials using hybridiser can lead to particle attrition. One of the distinct advantages of this technology is the cross application of the fundamental principles to develop solutions for a wide range of different problems. For instance understanding the role of surface texture of carrier particles on the strength of interaction between the guest and carrier particles can provide vital information on its impact on flowability, guest stability (as a composite particle) as well as functionality of the resultant particles. Research at Aston University within our group has led to the development of a dry particle coater which can produce micro functionalised particles. We have characterised the resultant particles using range of techniques including AFM, surface interferometry, confocal microscopy, inverse gas chromatography and particle size analysis. The resultant particles were studied for flowability enhancement, content uniformity and micro particle based modified drug release.
Rongshi Li
University of Nebraska Medical Center, USA
Title: Fragment-based, Structure-guided and Natural Product-derived Small Molecules as Antibiotic and Anticancer Agents

Biography:
Abstract:
Vakhtang Barbakadze
Tbilisi State Medical University, Georgia
Title: Caffeic Acid-Derived Biopolyether from Medicinal Plants as Anticancer Agent

Biography:
Vakhtang Barbakadze has completed his Ph.D and D.Sci. at the ages of 33 and 54 years from Zelinsky Instiute of Organic Chemistry, Moscow, Russia and Durmishidze Institute of Biochemistry and Biotechnology, Tbilisi, Georgia, respectively. He is the head of Department of Plant Biopolymers and Natural Products Chemical Modification at the Tbilisi State Medical University I.Kutateladze Institute of Pharmacochemistry. 1996 and 2002 he has been a visiting scientist at Utrecht University (faculty of pharmacy), The Netherlands, by University Scholarship and The Netherlands organization for scientific research (NWO) Scholarship Scientific Program, respectively. He has published more than 83 papers in reputed journals.
Abstract:
A new series of linear and regular caffeic acid-derived polyether, namely poly[3-(3,4-dihydroxyphenyl)glyceric acid] (PDPGA) was isolated and identified in the water-soluble, high molecular weight fractions obtained from Symphytum asperum, S.caucasicum, S.officinale, S.grandiflorum and Anchusa italica (Boraginaceae). According to data of 13C, 1H NMR, 2D 1H/13C HSQC experiment the polyoxyethylene chain is the backbone of the polymer molecule. The 3,4-dihydroxyphenyl and carboxyl groups are regular substituents at two carbon atoms in the chain. The repeating unit of this polymer is 3-(3,4-dihydroxyphenyl)glyceric acid residue. Most of the carboxylic groups of PDPGA from A. italica and S.grandiflorum unlike the polymer of S.asperum, S.caucasicum and S.officinale are methylated. The 2D DOSY experiment gave the similar diffusion coefficient for the methylated and non-methylated signals of A. italica PDPGA. Both sets of signals fell in the same horizontal. This would imply a similar molecular weight for methylated and non-methylated polymers. The synthesis of racemic monomer of PDPGA was carried out via asymmetric dihydroxylation of trans-caffeic acid derivatives using a potassium osmate catalyst and cinchona alkaloid derivatives as chiral auxiliaries. PDPGA and monomer exerted anti-cancer efficacy in vitro and in vivo against human prostate cancer (PCA) cells via targeting androgen receptor, cell cycle arrest and apoptosis without any toxicity, together with a strong decrease in prostate specific antigen level in plasma. However, our results showed that anticancer efficacy of PDPGA is more effective compared to its synthetic monomer. Overall, this study identifies PDPGA as a potent agent against PCA without any toxicity, and supports its clinical application.
Mariola Napiórkowska
Medical University of Warsaw, Poland
Title: Synthesis and cytotoxic properties of new derivatives of dicarboximides

Biography:
Mariola Napiórkowska is Ph.D on the Chair and Department of Biochemistry of Medical University of Warsaw. She has working at the Warsaw Medical University for 18 years. For many years she has engaged on the synthesis of compounds with biological activity. She is co-author of 23 publications and two patent applications.
Abstract:
The growth rate of the number of cancer cases is enormous and requires intensive research and introduction of new anticancer drugs. Ideally, these new drugs should possess improved pharmacokinetic parameters and high selectivity towards cancer cells providing less negative side effects. The present work describes the synthesis and cytotoxic activity of a large group of new derivatives of dicarboximides. In our previously studies with this group of compounds we have identified several derivatives that presented highest cytotoxicity to leukemia cells, lines K562, HL-60, respectively (IC50* in the range of 1-10µM). On the contrary, these compounds were non-toxic to non-tumor endothelial cells (HUVEC) and tumor adherent cells (HeLa). Taking into account the high activity of evaluated compounds, we decided to continue our work on the field of these derivatives. The main stage of synthetic works was the modification of the structure of dicarboximides in order to obtain products with improved solubility and bioavailability, while retaining their biological activity. In order to improve the solubility of the compounds hydrophilic groups such as -OH, -NH2, -NH-R were introduced to their structure. Thus we have synthesized 33 new derivatives. The structures of all new compounds were established by 1HNMR, 13CNMR and HR MS spectra. The obtained compounds were tested for their cytotoxic properties in cervix carcinoma (HeLa), chronic myelogenous leukemia (K562), acute lymphoblastic leukemia (MOLT-4) and normal endothelial (HUVEC) cells using MTT assay. In these screening studies we have identified 28 compounds that showed toxicity toward both HeLa, K562 and MOLT-4 cell lines.
Nadezhda Shchepina
Natural Sciences Institute of Perm State University, Russia
Title: Unusual Reaction of Phenylation and New Appoach for the Synthesis of Tritium Labeled Biomarkers with Quaternary Heterocyclic Nitrogen Atom

Biography:
Nadezhda Shchepina has completed her PhD at the age of 28 years from Leningrad (St. Petersburg) State University, Department of Radiochemistry. From 1972 year she works at the Laboratory of Radiochemistry of Perm State University (Scientist 1972-1980, Senior Scientist 1980-2002, Head of the Laboratory of Radiochemistry 2002-current). She was Visiting Associative Professor of Chemistry at Creighton University, Omaha, NE, USA (1978-1979). She defended the Doctoral Dissertation (Radiochemistry & Organic Chemistry) at Moscow M.V. Lomonosov State University in 2013 year. She is Full Professor at the Department of Organic Chemistry of Perm State University (2014-current). Nadezhda Shchepina has published more than 180 publications (24 patents of the USSR and 8 patents of Russin Federation, 85 articles in Russian and foreign reputed scientific journals).
Abstract:
A simple glance at databases of approved pharmaceuticals reveals the structural significance of nitrogen-based heterocycles in the drug design and engineering of pharmaceuticals, with nearly 60% of unique small-molecule drugs containing a nitrogen heterocycle. The pyridine ring can be considered one of the most simple, but at the same time one of the most important heteroaromatic structure. Many biological processes in the organism involve participation of compounds containing a pyridine structure, such as nucleic bases, ferments and enzymes. Moreover, conducted biological studies showed that the most promising and, in some cases, unique, are quaternary pyridinium derivatives. A major synthetic obstacle to accessing variously substituted pyridines is the fact that in the classical organic chemistry reaction of direct phenylation of the heterocyclic nitrogen atom is unknown and the quaternary aryl derivatives may be prepared only through a cyclization reaction. New features offers elaborated by us nuclear chemical method, namely, the use of the revealed reaction of direct heterocyclic nitrogen atom phenylation by nucleogenic (generated via tritium beta decay processes in tritiated benzene) free substituted and unsubstituted phenyl cations in order to obtain both inaccessible and unknown in the classical organic chemistry phenyl substituted six membered heterocyclic structures with quaternized nitrogen atom; one-step production of tritiumlabeled biomarkers with fixed tritium label in N-phenyl ring; and the study of pharmacological action of promising compounds with quaternary nitrogen scaffold by tritium label.
Gabriela Pricope
Centre of Advanced Research in Bionanoconjugates and Biopolymers, Romania
Title: Supramolecular G-quartet architectured hydrogels as drug delivery systems

Biography:
Supramolecular hydrogels have been extensively studied for their biomedical and pharmaceutical applications. Proprieties such as high water content and the soft consistency, similar to the consistency of natural tissue, contributes to their biocompatibility. Furthermore, the ability of different sized molecules to diffuse into (drug loading) and out of hydrogels (drug release) make these materials perfect candidates for drug delivery systems. In this report, hydrogels based on the formation of G-quartet were proposed and studied for the incorporating and releasing of the active compounds. An in situ method for drug loading was selected, thus the hydrogel network formulation and the drug encapsulation were accomplished simultaneously. The release of the encapsulated compounds was determined by diffusion in buffer solutions at physiological and acidic pH values. Drug-release measurements were performed using UV-Vis spectroscopy, fluorometry and circular dichroism spectroscopy. Presented work also shows the influence of structural components upon drug loading/release properties of the newly prepared hydrogels.
Abstract:
Gabriela Pricope is a biology graduate, with a master’s degree in microbial and cellular biotechnologies, currently PhD student in chemistry at ‘‘Petru Poni’’ Institute of Macromolecular Chemistry.
Alexander V. Sirotkin
Constantine the Philosopher University and Research Institute of Animal Production, Slovakia
Title: Plant molecules affecting female reproductive functions

Biography:
Prof. A.V. Sirotkin, PhD, DrSc is working as Professor at the Constantine the Philosopher University, as a Research Scientist at Research Institute of Animal Production in Nitra and as a Visiting Professor at the King Saud University in Ryiadh. He has about 600 publications including 120 full papers in the international journals. He is a member of editorial boards of 4 international journals and a recipient of more than 10 national and international awards.
Abstract:
The aim of our in vitro and in-vivo studies was to examine the potential influence of some medical and food plants and their constituents on ovarian functions and their potential usefulness as pharmacological stimulators of fecundity and protectors against the influence of environmental contaminants. For this purpose, we have study the influence of green tea, rooibos, ginkgo, flaxseed, yukka extracts, as well as of plant molecules resveratrol, curcumin, quercetin, daidzein, diosgenin on proliferation, apoptosis, release of hormones and response to gonadotropins of murine, porcine and rabbit ovarian cells as well as on rabbit fecundity. It was observed, that green tea, rooibos, ginkgo, flaxseed, extracts, as well as of resveratrol, curcumin, quercetin, daidzein, diosgeninare able to suppress proliferation, promote apoptosis, to alter the release of steroid hormones and to inhibit the response of cultured ovarian cells to hormonal stimulators FSH and IGF-I. On the other hand, some of these plants were able to prevent the action of environmental contaminants benzene, xylene and toluene on ovarian cells. Yucca extract expressed an opposite effect. Furthermore, feeding of rabbits with yucca and curcumin increased their fecundity. These observations suggest potential direct inhibitory influence of food and medical plants green tea, rooibos, ginkgo, flaxseed on ovarian functions. The similarity in plant and plant constituents effects suggest that the observed plant effects can be due to presence of curcumin, quercetin, daidzein and diosgenin. The potential anti-reproductive effect of these plants should be taken into account by their consummation. On the other hand, some plants or plant molecules could be used as stimulators of reproduction and fecundity and protectors against the influence of environmental contaminants.
Gevorg G. Danagulyan
Russian-Armenian University, Armenia
Title: Synthesis of heteroaryl-substituted pyrazolo[1,5-a]pyrimidines by recyclization of pyrimidinium salts

Biography:
Prof. Danagulyan has completed his PhD at the age of 27 from M. Lomonosov Moscow State University, in 2000 defended ScD thesis in Yerevan. Elected Corresponding Member of the National Academy of Sciences of Armenia (2010). Research areas: Chemistry of nitrogen-containing heterocycles and of biologically active substances. He has published more than 200 papers in reputed journals and is a member of editorial boards of a number of journals.
Abstract:
Compounds containing condensed pyrimidine systems and a bridged nitrogen atom are known to present interest as potential analgetics, antitumor drugs, bronchial spasmolytics and breathing stimulants. They form the composition of some drugs. We studied the reaction of 2-(ethoxycarbonyl)methyl-1,4,6-trimethylpyrimidinium iodide with hydrazides of C-pyridyl-, C-pyrimidinyl-, N-azolyl- and C-pyrazolyl-substituted carboxylic acids. This reaction was shown to result in recyclization and formation of ethyl 2-(pyrimidinylalkyl)- and 2-(azolylalkyl)-5,7-dimethylpyrazolo[1,5-a]pyrimidine-3-carboxylates. It is a new rearrangement of 1,2-dialkylpyrimidinium salts proceeding through the pyrimidine ring recyclization with inclusion of the nucleophilic reagent fragment into the transformation product. Heterocyclic acids hydrazides, in which a hydrazide fragment is immediately connected with a heterocyclic ring, also undergo a similar reaction. By the reactions of salt 1 with isonicotinic acid hydrazide (isoniazid) and pyrazol-3-carboxylic acid hydrazide we managed to obtain pyrazolo[1,5-a]pyrimidine derivatives that contained in position 2 pyridine and pyrazole rings. Besides pyrazolopyrimidines, the separation of reaction mixture provided in some cases also another recyclization product, 2-hydroxy-5,7-dimethylpyrazolo[1,5-a]pyrimidine.
Adriano Mollica
G. D’ annunzio†of Chieti-Pescara, Italy
Title: Newly potent opioid peptides with analgesic activity containing a xylene bridge

Biography:
Dr. Adriano Mollica is currently Associate Professor of Medicinal Chemistry at the Univ. “G. d’ Annunzio” of Chieti-Pescara, Italy and PhD in “Pharmaceutical Sciences. His research interests are focused on opioid peptides, neurodegeneration, treatment of chronic and neuropathic pain, multi-target compounds and nutraceuticals developments. He is co-author of 90 papers, one European Patent, two books and several oral communications. He is currently Editor in Chief of Current Bioactive Compounds and EBM for Mini Rev. in Med. Chem., Frontiers in chemistry (Medicinal and Pharmaceutical Chemistry), Prot. & Pept. Lett. and LDDD.
Abstract:
Novel cyclic analogues of enkephalin containing xylene bridge regioisomers (7a-c) have been synthesized and fully characterized as mixed m,d opioid receptors agonists.a,b The in vitro activity has been investigated showing a good affinity of 7a-c for both of them. In vivo biological assays revealed that 7b is the most potent cyclic analogue with the ability to maintain high level of analgesia after 60 and 90 minutes following i.c.v. and i.t. administrations respectively in Tail Flick test and long lasting analgesia after subcutaneous administration.
Hiroshi Maruta
PAK Research Center, Australia
Title: Development of PAK1-blockers (natural or synthetic) potentially useful for cancer therapy and longevity

Biography:
Ph.D. from University of Tokyo (in Pharmaceutical Sciences) in 1972. Research Associate at NIH (1973-80), Group leader at Max-Planck-Institute for Biochemie (1980-1984). Senior Scientist at Yale University and UCSD (1985-88). Laboratory Head at Ludwig Institute for Cancer Research (Melbourne Branch) (1988-2006). Visiting Professor at Hamburg University Hospital (2006-2007). Manager at PAK Research Center in Melbourne (2007-present).
Abstract:
PAK1 (RAC/CDC42-activated kinase 1) is the major oncogenic/ageing kinase that is responsible for a wide variety of diseases/disorders such as cancers, neurofibromatosis (NF), Alzheimer’s disease (AD), diabetes (type 2), hypertension, a variety of infectious and inflammatory diseases, epilepsy, schizophrenia, depression, autism, and obesity. In addition, PAK1 shortens the healthy lifespan (1), and is essential for PDGF-dependent melanogenesis as well (2), suggesting that PAK1-blockers could be elixirs and skin-whitening cosmetics. It should be worth noting that all herbal AMPK activators are PAK1-blockers as well as HSP (heat shock protein)-inducers, as the anti-oncogenic kinase LKB1 activates AMPK as well as inactivates PAK1, and activates HSP genes through the transcription factor FOXO that is activated by AMPK and inactivated by PAK1 (1). Thus, the potential market value of natural or synthetic PAK1-blockers/AMPK activators/ HSP-inducers would be huge, and pharmaceutical giants such as Pfizer, Roche, Novartis and Astrazeneca recently started developing potent PAK1-blockers. However, major problem of these synthetic PAK1-blockers for clinical application is their poor water-solubility and cell-permeability. Here we present a new trick for robustly boosting the anti-cancer activity and cell-permeability of several COOH-bearing PAK1-blockers without any loss of their water-solubility. According to Barry Sharpless (2001 Nobel-laureate), any COOH-bearing compounds could be esterized with water-soluble 1,2,3-Triazolyl alcohol through copper-catalyzed “Click Chemistry” (CC) in a high yield. Thus, in 2015, we have successfully synthesized 1,2,3-Triazolyl esters of two herbal PAK1-blcokers (artepillin C, ARC, and caffeic acid, CA) from propolis, and an old pain-killing PAK1-blocker (ketorolac) sold by Roche. The resultant esters (15A, 15C and 15K) are highly cell-permeable and their anti-cancer activity is 100 times of ARC, over 400 times of CA, and over 500 times of Ketorolac, respectively (3). Among these esters, 15K is the most potent anti-cancer/PAK1-blocker with IC50 =24 nM (against A549 lung cancer) and IC50=6 nM (against B16F10 melanoma) cells (3). Furthermore, 15K inhibits angiogenesis in ovo (fertilized eggs) with IC50 = 1 n mole/egg (Ahn, MK et al, unpublished observation). Currently we are testing if 15K extends the healthy lifespan of C. elegans, as does Okinawa propolis (OP) (4). Regarding OP and edible sea cucumber (SC), the major anti-cancer ingredients of OP and SC are geranylated flavonoids (Nymphaeols A-C), and a sulfated saponin called Frondoside A (FRA), respectively. We recently found that both Nymphaeols and FRA directly inhibit PAK1 in vitro with IC50 10 mM and 1 mM, respectively, in a selective manner (Nguyen, BC et al, unpublished observation).

Biography:
Abstract:
In this work we derive the generalized Hamilton–Jacobi equations for dynamical systems subject to nonholonomic constraints. The geometric formulation of Hamilton-Jacobi theory for such constraints is then developed, following the ideas of separation of variables technique and the canonical transformations. It is shown that the equations of motion which follow from the principle of d’Alembert are identical to the equations which follow from the variational action principle.
Bogdan Florin Craciun
Center of Advanced Research in Bionanoconjugates and Biopolymers, Romania
Title: Self-assembled polymeric vectors for gene delivery

Biography:
Bogdan Florin Craciun has obtained master degree at age 24 at Faculty of Chemistry, “Alexandru Ioan Cuza” University, Iasi, being specialized in organic chemistry. His work during master is highlighted by being co-author in three published papers, two of them represent the work accomplished in a practical stage made in Germany. Currently he is in first year of PhD at Petru Poni Institute of Macromolecular Chemistry, researching the field of drug delivering systems.
Abstract:
In recent decades a considerable attention was accorded to the field of drug delivery. A fraction of this domain is based on polymeric micelles with nanometer dimensions which proved their efficiency in vitro and in vivo testing. In this context, a library of self-assembled vectors based on squalene, benzene-1,3,5-tricarboxaldehyde (TA), branched polyethylenimine (PEI) and linear polyethylene glycol (PEG), was obtained as vectors for gene delivery. We have shown that this type of polymeric vectors possess a great affinity for binding the nucleic acid and good transfection efficiency on HeLa cell line. The main objective of the presented work was to find the best transfecting vector by tuning the ratios of components in the system, thus maximizing the transfection efficiency keeping a minimum grade of toxicity. We applied dynamic combinatorial chemistry for the preparation of vectors and it was proved the formation of polyplexes by gel electrophoresis and TEM. In vitro biological assessments show that the content of PEG in obtained polyplexes, plays a crucial role in transfection efficiency and cytotoxicity on HeLa cell line.
Khaldi A Mohammad
University of Tahri Mohamed Bechar, Algeria
Title: Antifungal activity of essential oil from Artemisia campestris L on Fungal Species Development

Biography:
Abstract:
This work studies the antifungal capacity of the essential oil of spontaneous aromatic plant with vocation medicinal used in the traditional treatments in the South-West of Algeria: Artemisia campestris L. The local plant tested gives a good essential oil yield (0.37%). The physico-chemical analysis of the essential oil of this plant specie has enables to us to even characterize to identify our oil. Antifungal activity of the essential oil was studied witch respect to seven fungal strains with various concentrations. The results of direct contact method show that the oil of Artemisia campestris L. is proven very effective on the mycelial growth of the moulds. All strains were inhibited at concentration as weak as 1/70 (v/v), Fusarium oxysporum f.sp. albedinis and Penicilluim expansum were most sensitive, being inhibited as from 1/800 (v/v) and 1/500 (v/v) respectively. This essential oil has a fungistatic effect. In addition to the growth of the mycelium, the essential oil of plant showed, in vitro, a antifungal activity at least important on the two other developmental stages, germination and the sporulation, of all fungi . All strains were inhibited at concentration as weak as 1/100 (v/v). Fusarium oxysporum f.sp. albedinis was most sensitive, being inhibited as from 1/1500 (v/v).
- final
- Drug Discovery and Development|Drug Designing Methodologies|Drug Delivery techniques

Chair
Catherine Guillou
University of Paris-Saclay, France

Co-Chair
Zeeshan Ahmad
De Montfort University, UK
Session Introduction
Shirly Kumala
Pancasila University Srengseng Sawah, Indonesia
Title: Isolation and Characterization Active Compound From Endophytic Fungi Onion Dayak {Eleutherineamericana (aubl.) Merr} as Antioxidant Activity

Biography:
Shirly Kumala has completed her PhD from Biomedic Faculty, University of Indonesia. Jakarta. She is the Dean of Pharmacy Faculty. University of Pancasila. Jakarta, Indonesia. She has published more than 25 papers in both international and national journals, and has been serving as a reviewer in Journal of Pharmacy.
Abstract:
Microorganisms, in particular, endophytic microbes have been well documented as alternative source of raw materials for drug development. Secondary metabolites produced by endophytic microbes have efficacious medicinal activity. The aims of this research focussed on isolation of the endophytic microbes from Bawang dayak leaf and evaluated their secondary metabolites. Isolation of endophytes were performed in PDA (Potato Dextrose Agar) using direct seed plant method. Endophytic fungi isolates with strongest antioxidant activity were fermented in PDY (Potato Dextrose Yeast) to produce large scale of the metabolites. Supernatant were extracted with ethyl acetate solvent. Ethyl acetate extract fractionated by column chromatography {SiO2, (i). chloroform –methanol = 50:1~10:1; (ii). Choloroform-Methanol = 5:1) and obtained three fractions. Further, colorimetric using free radical scavenger method was perform to assess their antioxidant activity. The highest antioxidant activity were identification by Nuclear Magnetic Resonance (1H- & 13C-NMR), infrared and mass spectroscopies showed the antioxidant compoud as flavonoid. In conclusion, the pure compound of secondary metabolites isolated from Bawang dayak leaf is flavonoid that could be responsible for its potent antioxidant activity.
Jim-Min Fang
National Taiwan University, Taiwan
Title: Phosphonate Congeners of Oseltamivir, Zanamivir and Peramivir as Effective Anti-Influenza Drugs

Biography:
Dr. Jim-Min Fang received his PhD degree in 1980 from Yale University. After finishing his postdoctoral research, he joined the Department of Chemistry, National Taiwan University (NTU), as Associated Professor in 1982. He is now an NTU Distinguished Professor and TBF Chair in Biotechnology. He also holds a joint appointment in the Genomics Research Center, Academia Sinica. His research interests are organic synthesis and chemical biology. He has published more than 250 papers.Â
Abstract:
Vaccination and drugs are effective for prevention and treatment of seasonal flu. However, drugs are especially needed in pandemic influenza before new vaccines can be produced. Influenza A is the most infectious type of influenza viruses. There are 18 subtypes of hemagglutinin (HA) and 11 subtypes of neuraminidase (NA). Avian influenza viral HA recognizes the 2,3-linked sialic acid receptor on the host cell surface, whereas human influenza viral HA recognizes the 2,6-linked sialo-glycoprotein receptors. NA is reponsible for breaking the connection between viral HA and the host cell, so that the progeny virus particle can be released to infect the surrounding cells. Pandemic influenza infection may occur due to the genetic reassortment of HA and NA. Inhibition of NA is thus a useful strategy in development of anti-influenza drugs. Zanamivir (RelenzaTM), oseltamivir (TamifluTM) and peramivir (RepiactaTM) are the NA inhibitors used for treatment of influenza. However, the on-market anti-influenza drugs still have shortcomings, such as the emergence of oseltamivir-resistance viruses, and non-oral availability of zanamivir and peramivir. In this presentation, we shall show the use of phosphonic acid as a bioisostere of carboxylic acid for developing more effective anti-influenza agents.Â

Biography:
Zeeshan completed his undergraduate and PhD (2007) from Queen Mary, University of London. He further conducted post-doctoral studies from Queen Mary and University College London for 6 years before taking up his first academic post as a Lecturer. Zeeshan is a full Professor and he is the EPSRC EHDA Network lead in the UK (a highly interdisciplinary initiative between academia and industry focusing on the development novel advanced drug delivery systems engineering). He is also a Royal Society Industry Fellow. He has published extensively in the area and his research group focuses on the development of novel drug delivery systems and their engineering platforms. He is on the editorial board for various notable journals and peer reviews for all major pharmaceutical and biomaterial journals.
Abstract:
Encapsulation is a crucial strategy in several aspects of Pharmaceutical development. This can be approached from both material and processing aspects and lends to controlled release, material safeguarding and providing several engineering benefits. This talk will look at encapsulation technologies from a processing perspective; specifically focusing on electrical encapsulation methods. An overview of the underlying process will be provided and examples of how such principles can be modulated to match emerging technologies (e.g. nanoparticles, 3D printing fibrous delivery systems) will be shown. Furthermore, the talk will also discuss briefly developments in the field and the emerging roles of industry for such technologies.
Franz-Josef Meyer-Almes
University of Applied Sciences Darmstadt, Germany
Title: E-BABE- Kinetic binding assays for the analysis of protein–ligand interactions

Biography:
Franz-Josef Meyer-Almes has completed his PhD at the age of 28 years from University of Goettingen. He has 10 years experiences in biotech and pharma companies. He is Professor for Physical Biochemistry and has published more than 40 papers in reputed journals and holds more than 10 patents and patent applications.
Abstract:
The importance of binding kinetics in terms of residence time and on-rate in drug discovery has been broadly accepted in the past few years. Furthermore, evidence has accumulated that the optimal binding mechanism of a drug to its target molecule is related to physiological efficacy as well as selectivity and thus drug safety. Homogeneous fluorescence-based binding assays have been shown to enable high throughput kinetics requiring only small amounts of protein and can be developed to elucidate even complex mechanisms of molecular recognition. A generalized approach is proposed that combines high quality kinetic and equilibrium data in an Integrated Global Fit analysis yielding the most probable binding mechanism.
Shaaban K. Mohamed
Manchester Metropolitan University, UK
Title: Utilizing eco-friendly nanoparticle techniques in improving drug delivery

Biography:
Shaaban K. Mohamed has completed his PhD at the age of 32 years from Minia University and postdoctoral studies from Didsburg University School of Chemistry, Germany and Manchester Metropolitan University, UK.. He is member of RSC and received the Knowlege Exchange award 2013, MMU, UK . He has published more than 260 papers in reputed journals and has been serving as an editorial board member of repute journals such as International Journal of Chemistry and Pharmaceutical Research, Greener Journal of Pharmacy and Pharmacology, and Journal of Pharmaceutical and Applied Chemistry.
Abstract:
The emergence of multidrug resistant (
Shaima El-Mowafi
National Research Centre, Egypt
Title: Synthesis and biological evaluation of novel antimicrobial cell-penetrating cyclic peptides

Biography:
Shaima El-Mowafi has completed her PhD from the Pennsylvania State University in 2014. She is currently a postdoctoral researcher at the National Research Centre in Egypt.
Abstract:
One of the greatest threats to human health in the twenty-first century is the development of antibiotic resistance. Gram-negative pathogens are remarkably successful in evading antibiotic action, due to their ability to maintain the integrity of their cell envelope. Recently, a cyclic octapeptide, named SI24, was identified from a genetic screen as an inhibitor of the σE cell envelope-sensing pathway, which is required for the virulence and viability in several Gram-negative pathogens. SI24 was found to be toxic to E.coli bacterial cells when expressed in vivo, but was not, however, an effective inhibitor when added exogenously, probably because it cannot cross the cell envelope (1). On the other hand, positively charged antimicrobial peptides have been considered as potential therapeutic sources of future generations of antibiotics for treating resistant pathogenic microbes because of their broad-spectrum activities. These candidates have been developed as cell-penetrating peptides (CPPs) and have become one of the emerging vehicles for delivery of cargo drugs. We thus hypothesize that conjugation of the cyclic peptide inhibitor SI24 with CPPs could enhance its permeability, and consequently its activity against bacterial cells, providing a paradigm for the development of antibiotics targeting a novel pathway in Gram-negative pathogens.
Matthew D. Lloyd
University of Bath, United Kingdom
Title: α-Methylacyl-CoA racemase (AMACR): Chemical biology approaches to novel prostate cancer drugs

Biography:
Matthew D. Lloyd graduated with a DPhil from Oxford on clavulanic acid biosynthesis. Following post-doctoral research at Brown University, U.S.A. and Oxford, he took up a lectureship at the University of Bath in 2002. He is currently Senior Lecturer (Associate Professor) in Pharmacy & Pharmacology. Research interests include prostate cancer, chemical biology, lipid metabolism, enzymes and inhibitors as drugs. He has published >70 research papers and is an editorial board member of The Journal Of Enzyme Inhibition and Medicinal Chemistry and The World Journal of Biological Chemistry. He is also a 4th Black Belt (International Instructor) in the Ch’ang-Hon Taekwon-Do.
Abstract:
α-Methylacyl-CoA racemase (AMACR; P504S) catalyses a key step in the degradation of branched-chain fatty acids and is important for the pharmacological activation of Ibuprofen and related drugs. Over-expression of AMACR correlates with tumorigenesis of many cancer types in particular, prostate cancer. Therefore, inhibition of AMACR is a promising chemotherapeutic strategy. Development of AMACR as a drug target has been hampered by the lack of a convenient biochemical assay for enzymatic activity, and therefore few inhibitors have been identified to date. We have developed a new, continuous colorimetric assay based on the elimination of 2,4-dinitrophenolate from a novel acyl-CoA substrate. Our fully developed enzyme assay can be performed in a high-throughput screening format using a microtitre plate. Our assay has been used to determine the kinetic parameters for the substrate, determine IC50 and Ki values for known inhibitors, reversibility of inhibition, and characterise irreversible inhibitors. IC50 values for ~30 known substrates and inhibitors were determined to reveal the first structure-activity relationship study against AMACR in which potency was related to the lipophilicity of the acyl-CoA side-chain. The most potent inhibitor was N-dodecyl-N-methylcarbamoyl-CoA (IC50 vs. AMACR = 400 pM). ‘High-throughput screening’ and IC50 determination of drug-like molecule libraries identified several new classes of inhibitors of AMACR. Our colorimetric assay now allows for screening and rational drug design approaches and full characterization of AMACR inhibitors as new agents against prostate cancer.
- Pharmaceutical Chemistry Novel Aspects|Pharmaceutical Analysis|Research Studies in Pharmacology

Chair
Xianglin Shi
Chemical Process R&D, Biogen Idec, USA

Co-Chair
Gervais Bérubé
Université du Québec à Trois-Rivières, Canada
Session Introduction
Tetsuo Narumi
Shizuoka University, Japan
Title: Peptidomimetic Study on Amyloid Fibril Formation by Alkene-type Dipeptide Isosteres

Biography:
Tetsuo Narumi has completed his Ph.D at the age of 28 years from Kyoto University with Prof. Nobutaka Fujii. He spent a year in US as a JSPS postdoctoral fellow with Prof. Jeffrey W. Bode at the University of Pennsylvania. In 2009, he began his academic career in Japan, at Tokyo Medical and Dental University with Prof. Hirokazu Tamamura. In 2013, he began his independent career at Shizuoka University, in Japan, as an associate professor in the Bioorganic Chemistry. He has published more than 50 papers in reputed journals and serving as a leading researcher in the fields of peptidomimetic science.
Abstract:
Alkene-type dipeptide isosteres have emerged as ideal ground state mimetics of scissile peptide bonds. Although halo-substituted alkenes such as fluoroalkene and chloroalkene have been thought as one of the promising surrogates of peptide bonds, the lack of suitable synthetic methods toward those isosteres has hampered their application to pharmaceutical chemistry. To address this issue, we have identified a multi-gram preparative and diastereoselective synthetic approach toward (Z)-chloroalkene dipeptide isosteres [(Z)-CADIs]. A key to our approach is the use of 1,4-asymmetric induction strategy in the organocuprate-mediated allylic alkylation of allylic gem-dichlorides adjacent to the chiral center that is extremely useful for the diastereoselective synthesis of (Z)-CADIs in high yields with excellent (Z)-selectivity and diastereoselectivity (Z/E = >20:1, >20:1 dr) [Narumi, T. et al. Org. Lett., 2015, 17, 2302.]. In this presentation, we will demonstrate the preparation of several peptidomimetics containing (Z)-CADIs by utilizing our synthetic approach, and explore the potentials of (Z)-CADIs as amide bond isosteres including the H-bonding ability of the (Z)-chloroalkene moiety, the tolerance to Fmoc-solid phase peptide synthesis conditions, and application to the peptides for amyloid fibril formation.
Xianglin Shi
Chemical Process R&D, Biogen Idec, USA
Title: Development of Novel Petasis Reaction Methodologies for Efficient and Scalable Synthesis of Biologically Interesting Compounds

Biography:
Abstract:
BIIB042, a g-secretase modulator, was pursued for its potential in the treatment of Alzheimer’s Disease. Med. Chem. synthesis consisted of four chemical reactions. A mixture of four diastereomers were made in the first step and carried through to the end of the synthesis. BIIB042 was obtained in ~4% overall yield via chiral HPLC separation. A seven-step process was developed for delivery of kilograms of BIIB042. A racemic mixture made in the first step was separated using SMB chromatography. Second chiral center was introduced in the last step by stereoselective hydrogenation reaction to afford BIIB042 in 95-96% ee. This route afforded the API in ~9% overall yield. Further improved synthesis included synthesis of 2 by Petasis reaction of enantioenriched aldehyde 1 followed by its conversion into BIIB042 in two steps (Scheme 1). Development of a mild and efficient Petasis reaction condition made synthesis of 2 in high yield without loss of ee possible. This new Petasis reaction condition was applicable to a variety of substrates (Shi, X, et al. J. Org. Chem. 2012, 77, 1154).
Johann F. Osma
Universidad de los Andes, Colombia
Title: Inclusion of divergent technologies in future point-of-care devices

Biography:
Dr. Johann F. Osma has completed his PhD at the age of 28 years in Chemical Engineering from Rovira i Virgili University. He is an Associate Professor at Universidad de los Andes (Colombia) and head of the biomicrosystems research field from the microelectronics research center (CMUA) at the same university. He manages a clean-room specialized on biosensors and microfluidic systems dedicated to environmental monitoring, hazardous material detection, defense and aerospace. Also, he is the general secretary of the Colombian Nanoscience Network.
Abstract:
Point-of-care devices have emerged as a viable solution for the monitoring and diagnosis of different pathologies without medical intervention, and are meant to provide non-trained individuals with real-time diagnostic results. In general, point-of-care devices use a biosensor and an end-user device for the detection of a target analyte, e.g. proteins, cells, nucleic acids and metabolites, which can be related to a specific disease. Although there are many different techniques through which an analyte can be detected, immunoassays and electrochemical measurements have shown to be of special interest. In particular, electrochemical based point-of-care devices measure a change in the charge transport capacity between two electrodes via cyclic voltammetry, or a change of the electrical impedance via electrochemical impedance spectroscopy. Hereby, the design and fabrication of two different electro-immuno point-of-care devices for the detection of the human papilloma virus (HPV) and tuberculosis are presented. In both cases, a well array is placed on gold electrodes were specific antibodies are immobilized. Each well behaves as an individual biosensor through which electrochemical measurements are used to determine the presence of the target protein. The biosensors are integrated with an electronics reader that constitutes the end-user device, and are meant to be used by non-trained
Nurmeilis
Syarif Hidayatullah State Islamic University. Indonesia
Title: Pharmacological Evaluation of Sedative and Hypnotic Activity of Ethyl p-methoxycinnamate and N-hydroxyethyl-p-cinnamamide

Biography:
Nurmeilis has completed her doctoral studies (Dr) at the age of 41 years from Universitas of Indonesia with majoring of pharmacy and work as a lecturer in pharmacy department, Faculty of medicine and health sciences, Syarif Hidayatullah State Islamic University and now as a Head of program study of pharmacy.
Abstract:
Ethyl p-methoxycinnammate (1) was found as a major isolated compound from the rhizome of Kaempferia galanga. This compound has been reported to have various biological activities such as mosquito repellent and larvicidal, anti-tuberculosis, sedative, anticancer, analgesic and anti-inflammatory and hypo pigmentary. In this research, we have evaluated the sedative and hypnotic activity of 1 and its amide derivative, N-hydroxyethyl-p-cinnamamide (2). The method was carried out by evaluating significant sedative and hypnotic effect of 1 and 2 at the doses of 100, 200 and 400 mg/kg (by oral route), compared to reference substance diazepam in hole board and thiopental-induced sleeping time methods. In conclusion, compound 1 has 2 has a potential sedative and hypnotic activity, whereas compound 1 was significantly different to the negative control (p≤0,05).
Azrifitria
Islamic State University (UIN) Syarif Hidayatullah Jakarta, Indonesia
Title: Spermicidal and Antifertility Activity of Costus Spiralis Leaf Ethanolic Extract in Male Rats

Biography:
Azrifitria has completed her PhD at the Indonesia University School of Medicine. She is the head of pharmacy department at islamic state University Syarif Hidayatullah Jakarta, Indonesia. She has published several papers in reputed journals and get some course at pharmacy field at tokushima bunry japan.
Abstract:
Costus spiralis belongs to costus genus which is one of the source of steroidal saponin that can potentially be an antifertility agent. This study investigated the effect of Costus spiralis leaf 70% ethanolic extract on male reproductive system using rat model. The study was divided into four groups of five adult male rats Sprague-Dawley aged 12 weeks; control group, and treatment group those received 12.5; 25 and 37.5 mg/kg body weight of Costus spiralis extract. The 70% ethanolic extract of Costus spiralis was administered orally once a day for 48 days. Different parameters were studied including sperm motility, sperm count, tubulus seminiferous diameter, spermatocyte pachytene at stage VII-VIII, serum testosteron and spermicydal activity. The result of the analysis showed that the extract dosages had a significant difference (p ≤ 0,05) on the decrease of sperm motility, tubulus seminiferous diameter, sperm count and number of spermatocyte pachytene at stage VII-VIII compared to control without commensurate decline in serum testosterone levels. The minimum effective concentration of Costus spiralis leaf 70% ethanolic extract to totally immobilise sperm within 20 seconds was 20 mg/mL. The results of present experiment suggested that the Costus spiralis leaf 70% ethanolic extract exerted a significant anti-spermatogenic effect in male rat.
Zilhadia
Syarif Hidayatullah Islamic State University, Indonesia
Title: Differentiation of Bovine and Porcine gelatin in vitamin C gummy by High Performance Liquid Chromatography using Principal Component Analysis

Biography:
Zilhadia has completed his PhD from Faculty of Pharmacy Universitas Indonesia. She is a lecturer in Pharmaceutical Chemistry and the director of Pharmacy Medicine Laboratory Syarif Hidayatullah State Islamic University since 2012 until now. She had joined the Syarif Hidayatullah State Islamic University since 2006. She has published several papers in reputed journals and becomes presenter in several international conferences.
Abstract:
Vitamin C gummy is one of vitamin favoured by Indonesia Children. Gelatin is an important component for the manufacture of vitamin C gummy that serves as a gelling agent. Gelatin can be obtained by hydrolysis of collagen derived from skin, connective tissue and bones of animals from both bovine and porcine. Gelatine from porcine is forbidden for Moslems and Jews. The aim of this study was to differentiate between bovine and porcine gelatine in vitamin C gummy by High Performance Liquid Chromatography (HPLC) combined with PCA. Vitamin C gummy was hydrolyzed by 6 n-hydrochloric acid, then derivatized using 6-amino quinolyl-n-hydroxysucsinimidil carbamat (AQC) and analyzed by reversed-phase HPLC. The HPLC spectra were analyzed using a chemometric method, principal component analysis (PCA), to classify both of gelatin. The results from PCA, which were subsequently represented by the Cooman’s plot showed a clear distinction between gelatin samples of bovine and porcine origins. This qualitative approach could determine the source of gelatin in food and pharmaceutical industries, especially in Vitamin C gummies.
- Pharmaceutical Chemistry Novel Aspects|Prospectives of Medicinal Chemistry | Heterocyclic Chemistry | Pharmaceutical Analysis

Chair
Mark McLaughlin
Merck Sharp & Dohme, USA

Co-Chair
Piotr Przybylski
Adam Mickiewicz University, Poland
Session Introduction
Azrifitria
Syarif Hidayatullah Jakarta, Indonesia
Title: Analysis of Cathinone and Cathine in Crimson and Green Catha edulis by GC-MS and Determination of The Spermicidal Activity

Biography:
Azrifitria has completed her PhD at the Indonesia University School of Medicine. She is the head of pharmacy department at islamic state University Syarif Hidayatullah Jakarta, Indonesia. She has published several papers in reputed journals and get some course at pharmacy field at tokushima bunry japan.
Abstract:
Catha edulis (khat) fresh leaves and tops are chewed or dried and consumed as tea. It is habitually chewed by many peoples in Yemen, East Africa and Indonesia. The abuse of Catha edulis (khat), widely, has been increasing since the mid-2000s. These substances are derivatives of the naturally occurring compound cathinone, which is the primary psychoactive component of khat. There were two types of khat leaves, that called crimson and green khat. The difference found in the branches which crimson khat had twigs maroon red and the green khat had brownish green. Viewing of cathinone and cathine levels used GC-MS because the components can be analyzed and characterized by exploiting the properties of their volatility. The efficiency for the extraction and isolation of cathinone and cathine were investigated. GC-MS chromatogram shown a cathinone content on green khat had a peak high 213,800, while the crimson khat had peak high 111,693 which means green khat contains more cathinone that had a retention time of about 22.3 minutes. The Chatine level on green and crimson khat was very low that had a retention time of about 22.7 minutes. The minimum effective concentration of the green and crimson khat khat extract to totally immobilise sperm within 20 seconds was 200 mg/ml. This Study shown that the extract of Catha edulis possesses spermicidal activity and cathinone level at a green khat more high than a crimson khat.
Piotr Przybylski
Adam Mickiewicz University, Poland
Title: Synthesis, antibacterial and anticancer potency of new lactone and lactam macrolide derivatives

Biography:
Dr. Piotr Przybylski, Chemistry Doctor's (Ph.D. - 2004) and Doctor Science (D. Sc.-2011) degrees at Adam Mickiewicz University in Poznan, now is an associate professor of organic chemistry, head of the research team at Faculty of Chemistry of Adam Mickiewicz University. He obtained „Maxima Cum Laude” award for best graduates of Faculty of Chemistry (2000) AMU Poznan, Stipend of President of Poznan city (2004), Award for Ph. D thesis of Prime Minister of Poland (2005), Award for Young Scientists (Warsaw 2005, 2006) and scholarship for research in West Pomeranian University of Technology of the Foundation for Polish Science (2007); postdoctoral researches in cooperation with Prof. F. Bartl from Charité – Universitätsmedizin, Institute Physik und Biophysik, Berlin and Humboldt Universität zu Berlin Lebenswissenschaftliche Fakultät Institut für Biologie, Berlin (2017, DAAD stipend), Germany. Currently Dr. Przybylski studies are focused on structure-activity relationships of natural products and their derivatives (macrolides, ionophore antibiotics and natural polyphenols) tautomerization, atropisomerization and proton transfer processes and the impact of these phenomenons on biological activity. Member of Ed. Board of Journal of Spectroscopy (since 2012) and Member of Committee of Chemical Sciences, Pol. Acad. Sci., PoznaÅ„ Division (since 2011). He has published 106 papers in reputed journals.
Abstract:
Macrolide antibiotics are large group of natural products of attractive biological properties and produced by Streptomyces strains. Macrolides can be classified via inter-alia the type and the size of the macrolide ring1 as for e.g. lactone macrolides 14-membered erythromycins, 15-membered azithromycins, and 16-membered spiramycins and leucomycins, or lactam 26-membered rifamycins as e.g. rifampicine. Macrolide antibiotics, especially lactone ones, can be divided also by the type of saccharide moiety attached at the aglycone.2 Mechanism of action of macrolide lactone antibiotics’ is based on the inhibition of bacterial protein biosynthesis at different stages by reversible binding at bacterial 50S subunit of ribosomes3 whereas macrolide lactam antibiotics mechanism of action as rifamycins depends on inhibition of bacterial RNA polymerases4. Our modifications were performed using cascade and ‘click’ approaches in aim to construct novel semisynthetic antibiotics of well-balanced physico-chemical parameters (lipophilicity, water solubility) and of improved docking mode at the biological target. For example, modifications at aglycone ring via complete reconstruction of saccharides parts using regio- and diastereoselective cascade combination of intramolecular esterifications, tandem E1cB eliminations and subsequent 1,2-addition to carbonyl followed by 1,6-conjugate addition at α,β,γ,δ – unsaturated aglycone yielded novel lactone macrolides of enhanced antibacterial and anticancer activities.5,6 We use also analogous combined cascade and ‘click’ approaches to modification of other group of natural macrolide antibiotics like lactone erythromycins to obtain alternatives to the currently used antibiotics in clinical therapy. The project is financially supported by Polish National Science Centre (NCN), decision number UMO-2015/19/B/ST5/00231.
Faizah S Aljohani
Taibah University, Saudi Arabia
Title: Hydrogen bonding interactions of tris and bis urea and thioureas with cyanide ligand in an analogue of the sub-site of [FeFe]-hydrogenase

Biography:
Faizah has completed his PhD from University of East Anglia. She is work under suprvised of Professor Chris Pickett . She has published two papers and three papers. Now, She work as Assistant Professor of Inorganic Chemistry at Taibah University in Saudi Arabia.
Abstract:
Second coordination sphere interactions of metal centres involving hydrogen bonding, ion-pairing or dipolar bonding can play an important role in determining the physical and chemical properties of molecular and biomolecular systems. The second coordination sphere interactions of coordinated cyanide in a model for the diiron subsite of [FeFe]-hydrogenase has been examined. The natural subsite in its protein environment catalyses hydrogen evolution (or uptake) at very fast rates ca 104 s-1 near the reversible potential of the H+ 1/2 H2 couple but such rates have yet to be attained in model systems. Controlling the electronic and geometric properties of synthetic diiron subsites by hydrogen bonding or other interactions might provide a means of attaining the turnover rates of the natural system. The specific approach taken in this work was to explore the potential binding properties of certain bis- and tris-(thio)ureas to the synthetic subsite analogue [Fe2(CO)4(CN)2pdt]2-. It is shown that hydrogen bonding interactions with the ligated cyanide using certain activated amides also perturbs spectroscopic and redox properties and importantly, the rate of protonation at the metal-metal bond and the stability of the resulting µ-hydride.
Yahdiana Harahap
Universitas Indonesia, Indonesia
Title: Method Development and Validation of Lercanidipine in Human Plasma by Liquid Chromatography Tandem - Mass Spectrometry

Biography:
Yahdiana harahap has completed her PhD from Department of Pharmacy, Institute Technology Bandung, Indonesia. Now she is the Head of Biavailability and Bioequivalence laboratory Faculty of Pharmacy, Universitas Indonesia. Prior to this position, she was the Dean of Faculty of Pharmacy, Universitas Indonesia. She has published more than 50 papers published in both International and National Journals. She has been invited to be the speakers in many international conference, especially in the field of BA/BE and Bioanalysis technique. She currently serves as an expert at Indonesia National Agency of Drug and Food Control, spesifically in BA/BE evaluation.
Abstract:
Lercanidipine is an antihypertensive of calcium channel blocker that is effective in treatment of patients with mild to moderate hypertension without affecting heart rate. The aim of this study was to obtain the optimum method and validate lercanidipine analysis in plasma using LC-MS/MS. Chromatographic separation was performed using Waters AcquityTM UPLC C18 1.7 µm (2.1 x 100 mm) column with mobile phase a mixture of 0.1% formic acid - methanol (20:80 v/v) with isocratic elution, the column temperature was 30°C with 0.2 mL/min flow rate and amlodipine as internal standard. Mass detection was performed on Waters Xevo TQD equipped with an electrospray ionization source in the MRM mode. Lercanidipine was detected at m/z 612.11 > 280.27 and amlodipine was detected at m/z 409.1 > 238.15. The optimum sample preparation was carried out by liquid-liquid extraction method using 5 mL mixture of n-hexane-ethyl acetate (50:50 v/v), vortexed for 3 mins, centrifuged at 4000 rpm for 20 mins, evaporated at 50°C for 30 mins, and reconstituted with 100 µL of mobile phase. This method was linear at concentration range of 0.025 to 10 ng/mL with r ≥ 0.9986, and fulfills the acceptance of accuracy and precision within and between run in three days. In addition, this method fulfills the acceptance criteria for selectivity, carry over, stability, dilution integrity and matrix effects based on Guideline on Bioanalytical Method Validation by the EMA in 2011.
Satoshi Mizuta
Nagasaki University, Japan
Title: Synthesis and Discovery of Highly Potent Agents, Trifluoromethyl Containing Heterocycles Against Influenza Virus

Biography:
Satoshi Mizuta received his Ph.D. at Nagoya Institute of Technology in 2008. He worked with Prof. Carlos. F. BarbasIII at The Scripps Research Institute. Then he went back to Japan to work at the Sagami Chemical Research Institute in 2010-2011. He joined the VG group as a Marie-Curie Fellow in September 2011 until May 2013, to work on the catalytic trifluoromethylation and the development of synthetic method for [18F]labelling. Satoshi is working at Nagasaki University, Japan. His current research interest is the drug design for antivirus.
Abstract:
Trifluoromethyl heterocycles have been an important motif of pharmaceutical drugs and agrochemicals because the presence of a CF3 group can cause the improved metabolic stability, lipophilicity and bioavailability. Nowadays, numerous CF3 substituted heterocycle-containing pharmaceuticals on the market can be witnessed, with examples such as trifluridine, efavirenz, celecoxib and mefloquin. Over the past decades, there has been an increasing interest in the development of method for the efficient synthesis of such fluorinated heterocyclic molecules as potential biological targets. Nevertheless, synthetic method accessing an array of CF3-containing heterocycles remains underdeveloped, in particular for nonaromatic heterocycles. Considering the difficulty of introducing CF3 moiety in nonaromatic ring systems, divergent synthesis using a simple and readily available CF3-containing precursor to convert into the diverse set of trifluoromethyl heterocyclic compounds may be one of the versatile and straightforward strategies for drug discovery. In this study, a series of α-trifluoromethyl α,β-unsaturated lactones and trifluoromethyl pyrazolinones were designed and synthesized. These two structural motifs of trifluoromethyl heterocycles were synthesized based on the synthetic pathway including a tandem stereoselective functionalization of the key precursor 3,3-dibromo-2-trifluoromethyl acrylic acid ethyl ester and intramolecular cyclization reaction. Further modification by Suzuki-Miyaura cross-coupling reaction provided a set of multi-functionalized α,β-unsaturated lactones and pyrazolinones. All synthesized compounds were bioassayed in vitro to determine their inhibitory activity against influenza A virus. We further modified a few potent hits in a viral inhibition assay and cunducted SAR studies on these scaffolds. The results showed that CF3-containing spirolactones possessed promising inhibitory activity being nearly active as oseltamivir.